Indicating that the prescribed dose of zanamivir is not sufficient to inhibit bacterial neuraminidases. Therefore, if a certain amount of neuraminidase activity, originating from bacteria, is present on the surface of the respiratory tract, influenza virus infection, release and spread may not be suppressed by NA inhibitor drugs. In agreement with this possibility, it has been reported that receiving professional oral care and oral health guidance from a dental hygienist reduces both the number of oral bacteria and the activities of neuraminidase in saliva, resulting in a reduction in the risk of infection from influenza. Altogether, the control of bacterial neuraminidases in the upper respiratory tract should be taken in consideration when using prescribed NA inhibitors in order to minimize reduced drug potency. Both of these domains recognize the phospho-Ser/Thr-Pro bonds present in mitotic phosphoproteins. Pin1 is distinct from two other PPIase families, cyclophilin and FK506 binding protein, since Pin1 only has PPIase activity for phosphorylated substrates. Pin1 catalyzes prolyl cis-trans isomerization to function as a FDA-approved Compound Library molecular timer regulating the cell cycle, cell signaling, gene expression, immune response, and neuronal function. Pin1 is overexpressed in many cancer lines, and plays an important role in oncogenesis. Because of its significant role in cell cycle regulation by a unique mechanism, Pin1 represents an intriguing diagnostic and therapeutic target for cancer. Several promising classes of Pin1 inhibitors have been synthesized as potential lead compounds, including designed inhibitors, and natural products. The mechanisms of the PPIases, cyclophilins and FKBPs, were shown to go through a twisted amide transition state. Evidence included secondary deuterium isotope effects, molecular modeling, mutagenesis, and bound inhibitor structure. We anticipated that the ketones would be poor 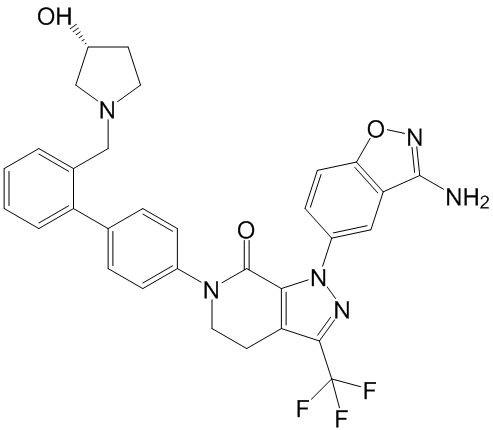 inhibitors, while the reduced amides, as twisted-amide analogues, would fare better. Indeed, the reduced amide 3 is a better Pin1 inhibitor than a similarly substituted substrate analogue -alkene isostere 5. Our crystal structure of reduced amide 4 bound to the Pin1 catalytic site adopted a trans-pyrrolidine conformation, supporting the twisted-amide mechanism. Ketones have been widely used as analogues of aldehydes or carboxylic acids to inhibit serine, cysteine, and aspartyl proteases. Substrate-analogue ketones have not yet been Dabrafenib developed as inhibitors of Pin1. Juglone is a ketone natural product that was shown to be a non-specific inhibitor of Pin1 through Michael addition to a surface Cys thiol of Pin1, resulting in unfolding. Daum et al developed a series of aryl indanyl ketone inhibitors of Pin1; the best inhibitor had an IC50 value of 0.2 mM. These inhibitors were reversible and cell penetrating, and they showed biological activities against p53 and b-catenin. Daum et al proposed that the aryl indanyl ketones mimic the transition state of the twisted amide, based on the conformation in a crystal structure. a-Ketoamides 6a and 6b were designed as potential transition state analogue inhibitors of Pin1, but their weak inhibition could not be used support either the twisted-amide or the nucleophilic-addition mechanism. As important targets for b-lactams and against the background of increasing clinical resistance to existing antibiotics, much effort has been directed toward developing new inhibitors of PBPs. Some approaches have sought to adapt the b-lactam moiety by, for instance, incorporating elements of the peptide substrates onto one of the R1 or R2 side chains, while others have synthesized compounds that mimic the tetrahedral intermediates of the reaction, including phosphonates and boronates.
inhibitors, while the reduced amides, as twisted-amide analogues, would fare better. Indeed, the reduced amide 3 is a better Pin1 inhibitor than a similarly substituted substrate analogue -alkene isostere 5. Our crystal structure of reduced amide 4 bound to the Pin1 catalytic site adopted a trans-pyrrolidine conformation, supporting the twisted-amide mechanism. Ketones have been widely used as analogues of aldehydes or carboxylic acids to inhibit serine, cysteine, and aspartyl proteases. Substrate-analogue ketones have not yet been Dabrafenib developed as inhibitors of Pin1. Juglone is a ketone natural product that was shown to be a non-specific inhibitor of Pin1 through Michael addition to a surface Cys thiol of Pin1, resulting in unfolding. Daum et al developed a series of aryl indanyl ketone inhibitors of Pin1; the best inhibitor had an IC50 value of 0.2 mM. These inhibitors were reversible and cell penetrating, and they showed biological activities against p53 and b-catenin. Daum et al proposed that the aryl indanyl ketones mimic the transition state of the twisted amide, based on the conformation in a crystal structure. a-Ketoamides 6a and 6b were designed as potential transition state analogue inhibitors of Pin1, but their weak inhibition could not be used support either the twisted-amide or the nucleophilic-addition mechanism. As important targets for b-lactams and against the background of increasing clinical resistance to existing antibiotics, much effort has been directed toward developing new inhibitors of PBPs. Some approaches have sought to adapt the b-lactam moiety by, for instance, incorporating elements of the peptide substrates onto one of the R1 or R2 side chains, while others have synthesized compounds that mimic the tetrahedral intermediates of the reaction, including phosphonates and boronates.
Month: August 2019
With some additional protein structure information was built and a ligand-based approach was followed instead
The inhibitor design concept of the present study triggered the synthesis of compounds 6 and 21 as promising new 17b-HSD1 inhibitors by optimizing a novel, in silico identified, core scaffold. The classical medicinal chemistry approach of rigidification was successfully applied to compound 5 and led to the discovery of the highly potent benzothiazole 6. The introduction of the aromatic benzothiazole freezes the position of hydroxy group in an ideal position to establish an H-bond with H221. In addition, this aromatic benzothiazole can undergo a cation-p interaction with Arg258, explaining the high gain in KRX-0401 potency of 6 compared to 5. In the optimization process the carbonyl bridge of 6 was varied using several linkers with different lengths, geometries and Hbonding properties. From the biological results as well as from the performed in silico studies it became apparent, that the 17b-HSD1 inhibitory activity is highly influenced by the nature of the linker: the comparison of inactive compounds showing a tetrahedral bridge geometry with the active, planar carbonyl and amide derivatives led us to conclude that a flat geometry of the linker is required for activity. The fact that the retroamide 21 is five times more active than the amide 18 can be explained by a steric clash observed between the carbonyl of amide bridge and Leu149. Furthermore, the carbonyl group of 21 was found to establish an H-bond interaction with Tyr218 which is not possible for 18. Comparing the binding modes of 6 and 21, it becomes apparent that the hydroxyphenyl moieties of the two compounds do not interact with the same area of the enzyme. In the case of compound 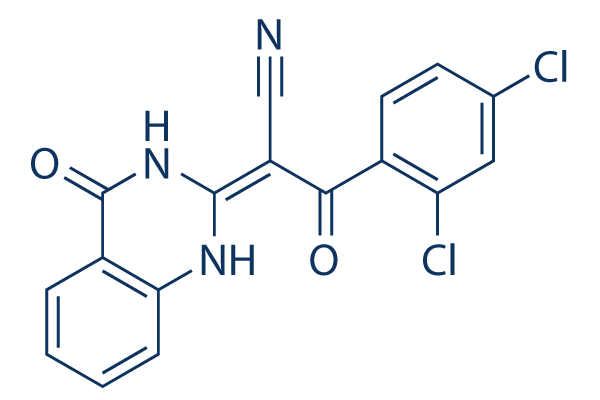 6, HY5 and D4 are plausible features covered by the hydroxyphenyl moiety. The meta-hydroxyphenyl moiety of 21, on the other hand, exploits HY1 and AD1. The difference in activity between 6 and 21 is in SB203580 agreement with the number of features covered by each compound. It is striking that the newly discovered class of benzothiazole derivatives shows structural characteristics which are similar to those of other classes of 17b-HSD1 inhibitors: two phenolic hydroxy-groups separated by a rather unpolar scaffold structure. The necessity for the lipophilicity of the scaffold is reflected by the gain in potency observed with the thiourea compared to the less lipophilic urea. The analysis of the amino acid residues which surround compound 6 in its pharmacophore binding pose indicates that two hydrogen bonds with Asn152 and one p-p interaction with Tyr155 are established. Recently published docking studies suggest similar interactions for bicyclic substituted hydroxyphenylmethanones. Interestingly, there is a decrease of activity in both compound classes when the hydroxy group is shifted from the meta- to the para position. This similarity in SAR supports the hypothesis that the hydroxyphenyl moieties of both compound classes bind in the same area of the enzyme. In order to evaluate the protein-ligand interactions, the ligands of the different X-ray structures studied were replaced by compounds 6 and 21 according to their pharmacophoric binding modes and the interactions between the inhibitors 6 and 21 and each of the crystal structures were examined. The maximum number of interactions was observed with the crystal structure 1equ, originally containing the inhibitor equiline. The reason for this is the residue Arg258 which protrudes into the active site in case of 1equ. The importance of this amino acid residue was already postulated by Alho-Richmond et al., who proposed to target it in the inhibitor design process. The biological assays employed for the evaluation of inhibitory potency towards 17b-HSD1 and 2 use well established conditions.
6, HY5 and D4 are plausible features covered by the hydroxyphenyl moiety. The meta-hydroxyphenyl moiety of 21, on the other hand, exploits HY1 and AD1. The difference in activity between 6 and 21 is in SB203580 agreement with the number of features covered by each compound. It is striking that the newly discovered class of benzothiazole derivatives shows structural characteristics which are similar to those of other classes of 17b-HSD1 inhibitors: two phenolic hydroxy-groups separated by a rather unpolar scaffold structure. The necessity for the lipophilicity of the scaffold is reflected by the gain in potency observed with the thiourea compared to the less lipophilic urea. The analysis of the amino acid residues which surround compound 6 in its pharmacophore binding pose indicates that two hydrogen bonds with Asn152 and one p-p interaction with Tyr155 are established. Recently published docking studies suggest similar interactions for bicyclic substituted hydroxyphenylmethanones. Interestingly, there is a decrease of activity in both compound classes when the hydroxy group is shifted from the meta- to the para position. This similarity in SAR supports the hypothesis that the hydroxyphenyl moieties of both compound classes bind in the same area of the enzyme. In order to evaluate the protein-ligand interactions, the ligands of the different X-ray structures studied were replaced by compounds 6 and 21 according to their pharmacophoric binding modes and the interactions between the inhibitors 6 and 21 and each of the crystal structures were examined. The maximum number of interactions was observed with the crystal structure 1equ, originally containing the inhibitor equiline. The reason for this is the residue Arg258 which protrudes into the active site in case of 1equ. The importance of this amino acid residue was already postulated by Alho-Richmond et al., who proposed to target it in the inhibitor design process. The biological assays employed for the evaluation of inhibitory potency towards 17b-HSD1 and 2 use well established conditions.
VEGFR-2 kinase inhibitor that progressed to Phase III clinical trials for metastatic colorectal roliferative activity
Agonists of the aryl hydrocarbon receptor have been of interest to the pharmaceutical industry for many years. This interest originally stemmed from the observation that the AHR is a ligand-activated transcription factor that regulates the adaptive metabolism of xenobiotics and because receptor XL-184 binding is a known step in the carcinogenic and toxic action of environmental pollutants like 2,3,7,8-tetrachlorodibenzo-p-dioxin. Thus, agonism of the AHR has commonly been considered a signature for drugs that upregulate phase-I and phase-II metabolic systems and also for chemicals with pharmacological similarity to a known human carcinogen. As a result, AHR agonism has largely been considered a hazard signature for environmental chemicals and drugs in the pharmaceutical pipeline. Recent insights related to the normal physiological role of the AHR are changing our view of receptor agonism to one where agonism might be considered to hold therapeutic value. A number of recent reports are identifying new biological processes that might be influenced by endogenous receptor ligands. For example, descriptions of mice harboring a null allele at the Ahr locus indicate that receptor signaling plays an important role in normal cardiovascular development and function. The therapeutic potential related to this biology is demonstrated by the observation that potent AHR agonists like TCDD can correct developmental aberrations in hepatic blood flow under conditions of AHR hypomorphism. More recently, a role for the AHR in immunology has been emphasized by reports that activation of this receptor with ligands, such as TCDD, can lead to the generation of regulatory T-cells, while activation with other ligands, such as formylindolocarbazole can lead to Th17 cell formation. The potential clinical importance of this finding is supported by the observation that TCDD is able to ameliorate the symptoms of experimental autoimmune encephalomyelitis in mice, whereas FICZ aggravates this syndrome. Additional studies have supported the idea that Axitinib VEGFR/PDGFR inhibitor ligands can play a role in improving allograft acceptance after transplantation. The importance of the AHR in immunology has also been extended by a series of papers demonstrating the central importance of this receptor in the presence and maintenance of intraepithelial lymphocytes and lymphoid tissue inducer cells in the gut, highlighting that the AHR and its ligands play a role in normal physiology of the immune system and response to the outside environment. We have begun a search for agonists and antagonists of the AHR as part of an effort to develop a new class of receptor ligands with therapeutic potential for the treatment of vascular or immunological disease. Our initial strategy is to screen compounds that are pharmacologically well studied and that pose less environmental or health risks as compared to TCDD. Our approach to initially screen a library of compounds with known biological activity was chosen for three reasons. First, well studied compounds hold greater probability of prior toxicological and pharmacological characterization and thus may move into clinical  settings more quickly. Second, identification of AHR ligands in classes of pharmacologically active compounds already in the clinic could shed additional insights into their mode of action, as well as identify compounds with understandable offtarget effects. Third, pharmacological information about novel AHR agonists could provide insight into the endogenous mechanism of action of this receptor or reveal the biological pathways in which the receptor participates during development.
settings more quickly. Second, identification of AHR ligands in classes of pharmacologically active compounds already in the clinic could shed additional insights into their mode of action, as well as identify compounds with understandable offtarget effects. Third, pharmacological information about novel AHR agonists could provide insight into the endogenous mechanism of action of this receptor or reveal the biological pathways in which the receptor participates during development.
Comparison of the ability of IKK family members to phosphorylate these different peptide substrates
The phosphorylation motif for TBK1 has not been previously reported. Here, a positional scanning peptide library technology was used to determine the optimal phosphorylation motif for TBK1. We demonstrate that the substrate specificity of TBK1 is identical to that of IKKe, but differs from the phosphorylation motif of IKKb at key positions. Importantly, we also demonstrate that, like IKKe, TBK1 phosphorylates its predicted optimal peptide more efficiently than an optimal peptide for IKKb or a peptide containing the IKKb phosphorylation sites present in IkBa. We then used this information to develop and validate an IKKe/TBK1 peptide substrate appropriate for highthroughput chemical screening and executed a high-throughput screen against both TBK and IKKe. The development of effective small-molecule screening technologies for kinases is dependent on appropriately measuring changes in enzyme activity. While phosphorylation of a known protein substrate can be measured as a reporter for kinase activity, a peptide substrate is usually superior, as it is easier to generate large, consistent quantities, and is  more amenable to the development of non-radioactive assays. However, the generation of an optimal peptide substrate requires a thorough understanding of kinase substrate specificity, and this information is only available for a small fraction of the.500 protein kinases in the human genome. The substrate specificities of three IKK family members, IKKa, IKKb and IKKe, have recently been described. Like IKKe, TBK1 is a noncanonical IKK family member which regulates Type I interferon signaling and may play a role in oncogenesis. Here, a positional scanning peptide library technology was utilized to identify the optimal phosphorylation motif for TBK1. The substrate specificity of TBK1 is identical to that of related kinase IKKe. Interestingly, the substrate specificities of the noncanonical IKKs share overlapping characteristics with the substrate specificity of the canonical IKKs, but the optimal peptide substrates for these kinases are quite different. These data allowed the generation of a peptide substrate for TBK1 and IKKe which is amenable to high-throughput screening. This technology was then used to screen the LOPAC library and a kinase-focused library to discover in vitro inhibitors of TBK1 and IKKe. This HTS revealed that 227 compounds in this library inhibited TBK1 at a concentration of 10 mM and 57 compounds inhibited IKKe, including several compounds that inhibited these enzymes at sub-micromolar concentrations. Of the compounds tested in this screen, the molecules in the LOPAC library were of MDV3100 particular interest since this library contains known bioactive molecules. The best TBK1/IKKe inhibitors from the LOPAC library are therefore shown in Table S1. Unfortuntately, none of the compounds from the LOPAC library were among the best inhibitors of IKKe or TBK1, and many lacked specificity as they also inhibited IKKa. Studies examining the ability of the compounds in Figure 7 to inhibit TBK1 or IKKe in cell-based assays are ongoing. As TBK1 and IKKe are points of convergence for both inflammatory and oncogenic signaling pathways, the further refinement of novel TBK1/IKKe inhibitors may provide NVP-BKM120 cost powerful new therapeutic drugs for inflammatory disorders or cancer. The lipid droplet is a subcellular structure that exists in a range of organisms from archaea to mammals. The LD used to be regarded as an inert lipid depot, but recent studies have revealed that it is an active organelle engaged in a wide range of activities. The main function of LDs is to store lipids and to supply them for various cellular needs, such as b-oxidation, membrane biogenesis, and lipoprotein synthesis.
more amenable to the development of non-radioactive assays. However, the generation of an optimal peptide substrate requires a thorough understanding of kinase substrate specificity, and this information is only available for a small fraction of the.500 protein kinases in the human genome. The substrate specificities of three IKK family members, IKKa, IKKb and IKKe, have recently been described. Like IKKe, TBK1 is a noncanonical IKK family member which regulates Type I interferon signaling and may play a role in oncogenesis. Here, a positional scanning peptide library technology was utilized to identify the optimal phosphorylation motif for TBK1. The substrate specificity of TBK1 is identical to that of related kinase IKKe. Interestingly, the substrate specificities of the noncanonical IKKs share overlapping characteristics with the substrate specificity of the canonical IKKs, but the optimal peptide substrates for these kinases are quite different. These data allowed the generation of a peptide substrate for TBK1 and IKKe which is amenable to high-throughput screening. This technology was then used to screen the LOPAC library and a kinase-focused library to discover in vitro inhibitors of TBK1 and IKKe. This HTS revealed that 227 compounds in this library inhibited TBK1 at a concentration of 10 mM and 57 compounds inhibited IKKe, including several compounds that inhibited these enzymes at sub-micromolar concentrations. Of the compounds tested in this screen, the molecules in the LOPAC library were of MDV3100 particular interest since this library contains known bioactive molecules. The best TBK1/IKKe inhibitors from the LOPAC library are therefore shown in Table S1. Unfortuntately, none of the compounds from the LOPAC library were among the best inhibitors of IKKe or TBK1, and many lacked specificity as they also inhibited IKKa. Studies examining the ability of the compounds in Figure 7 to inhibit TBK1 or IKKe in cell-based assays are ongoing. As TBK1 and IKKe are points of convergence for both inflammatory and oncogenic signaling pathways, the further refinement of novel TBK1/IKKe inhibitors may provide NVP-BKM120 cost powerful new therapeutic drugs for inflammatory disorders or cancer. The lipid droplet is a subcellular structure that exists in a range of organisms from archaea to mammals. The LD used to be regarded as an inert lipid depot, but recent studies have revealed that it is an active organelle engaged in a wide range of activities. The main function of LDs is to store lipids and to supply them for various cellular needs, such as b-oxidation, membrane biogenesis, and lipoprotein synthesis.
PRL-3 first gained notoriety as a marker for metastasis when the Vogelstein lab found PRL-3 levels high
For this reason, the protective effect of CQ in the murine EBOV challenge model is encouraging. None of the reported studies address the pharmacodynamics of the antiviral activity by demonstrating that the compound accumulates in the relevant tissue or compartments where the virus is replicating in vivo. Chloroquine has a large volume of distribution, which Temozolomide suggests that its rapid dissemination into extravascular tissues may impact its inhibitory activity. Clearly, the spectrum of viruses for which this class of compounds would be Reversine useful in vivo will be strongly determined by this factor, as well as by the potency of the compound itself in inhibiting specific steps in viral replication. Improvements in formulation, such as encapsulation within liposomes may also be of utility in modifying the pharmacokinetics of  CQ in vivo. The antiviral activity of CQ may serve as an initial starting point for antiviral development through optimization of the 4AQ scaffold and by exploiting the decades of experience in toxicological investigation for this class of compounds. Significant effort has been expended in optimizing derivatives of CQ for malaria strains that have acquired resistance. By optimizing the antiviral activity of these compounds for short- or intermediate-term therapeutic dosing, it should be possible to develop analogs with entirely different properties than those required for antimalarial activity, including lower toxicity. We have successfully identified many clinically useful drugs that are potential inhibitors of bacteria and virus infection. The efficacy of lomefloxacin against BA and CQ against EBOV in vivo has not been previously reported. The ability of erythromycin to inhibit filoviruses as well as bacteria is intriguing and suggests that this drug can act not only by impacting bacterial growth but also on the cell itself, possibly by altering uptake of the pathogen. Many other pathogen-specific drugs were identified that will require evaluation in animal models. The identification of these compounds lends credence to the repurposing approach for novel drug discovery against high containment and/or biodefenserelated pathogens. The potential to reduce the time from bench to clinic is great, and accelerating this process would save lives in the event of an outbreak of any pathogen. In the past decade, Phosphatase of Regenerating Liver family members have been touted as molecular markers that significantly correlate to the ability of cancers to metastasize,,. In addition, laboratory studies indicate that PRLs are promising therapeutic targets; interfering with PRL function using antibodies and RNA interference has shown dramatic reduction in tumor formation in mice,. PRL-1 was first isolated as a novel tyrosine phosphatase that is immediately transcribed following a partial hepatectomy, continually expressed in a number of tumor cell lines and able to transform non-tumorigenic cells,. Later, PRL-2 and PRL-3 were identified by sequence analysis. Studies in cell culture indicate that exogenous expression of PRLs can induce cell proliferation,,,, migration,,, and invasiveness,,. Most significantly, constitutive expression of PRL1 and -3 enable cultured cells to form tumors when injected into mice,,. The potential of increased levels of PRLs to actively contribute to oncogenesis complements dozens of studies correlating PRL expression to tumor aggressiveness.
CQ in vivo. The antiviral activity of CQ may serve as an initial starting point for antiviral development through optimization of the 4AQ scaffold and by exploiting the decades of experience in toxicological investigation for this class of compounds. Significant effort has been expended in optimizing derivatives of CQ for malaria strains that have acquired resistance. By optimizing the antiviral activity of these compounds for short- or intermediate-term therapeutic dosing, it should be possible to develop analogs with entirely different properties than those required for antimalarial activity, including lower toxicity. We have successfully identified many clinically useful drugs that are potential inhibitors of bacteria and virus infection. The efficacy of lomefloxacin against BA and CQ against EBOV in vivo has not been previously reported. The ability of erythromycin to inhibit filoviruses as well as bacteria is intriguing and suggests that this drug can act not only by impacting bacterial growth but also on the cell itself, possibly by altering uptake of the pathogen. Many other pathogen-specific drugs were identified that will require evaluation in animal models. The identification of these compounds lends credence to the repurposing approach for novel drug discovery against high containment and/or biodefenserelated pathogens. The potential to reduce the time from bench to clinic is great, and accelerating this process would save lives in the event of an outbreak of any pathogen. In the past decade, Phosphatase of Regenerating Liver family members have been touted as molecular markers that significantly correlate to the ability of cancers to metastasize,,. In addition, laboratory studies indicate that PRLs are promising therapeutic targets; interfering with PRL function using antibodies and RNA interference has shown dramatic reduction in tumor formation in mice,. PRL-1 was first isolated as a novel tyrosine phosphatase that is immediately transcribed following a partial hepatectomy, continually expressed in a number of tumor cell lines and able to transform non-tumorigenic cells,. Later, PRL-2 and PRL-3 were identified by sequence analysis. Studies in cell culture indicate that exogenous expression of PRLs can induce cell proliferation,,,, migration,,, and invasiveness,,. Most significantly, constitutive expression of PRL1 and -3 enable cultured cells to form tumors when injected into mice,,. The potential of increased levels of PRLs to actively contribute to oncogenesis complements dozens of studies correlating PRL expression to tumor aggressiveness.