The present study examined the roles of NaB and TSA on several parameters, biochemical and morphological, of the H460 cell line of lung cancer cells in order to clarify how these HDACis interferes with tumor cell homeostasis. The data showed conclusively that treatment with NaB for 24 h lead to a generally enhanced oxidative metabolism clearly suggesting that HDACis may transcend their canonical role at the chromatin level. Whether sporadic or hereditary, most if not all types of cancer ultimately derive from single cells that have undergone irreversible biochemical reprogramming. The phenotypes acquired by the clones of transformed cells are such that the intrinsic pathways normally acting as safeguards for the tissue and the organism become subverted and/or abrogated. The phenotype that confers virtually limitless replication to the transformed cells is costly in terms of energy. In rapidly growing BAY 43-9006 Raf inhibitor tumors, the prevalent anabolism must be accompanied by upregulated pathways that ultimately increase the rate of ATP synthesis for all processes connected to growth and invasiveness and hence necessarily involve elements of the intermediary metabolism. To make matters even more complex, it is known that the metabolic reprogramming exhibited by transformed cells is not homogeneously distributed throughout the tumor. Cells located at the centre of the tumor mass are under more severe anoxic conditions than those at the periphery and consequently two or more populations are formed that can be loosely classified as aerobic and anaerobic tumor cells depending on their location in this O2 gradient. Within a tumor, the mixed cell population of hypoxic and normoxic cells exchange metabolites between each other establishing a network of Gefitinib side effects complementary pathways that collectively have been termed biochemical symbiosis. In this situation it can be inferred that mitochondria of at least part of the cell population are functional. In the present paper we confirmed that aerobic glycolysis and oxidative metabolism coexist in tumor cells and most likely complement each other through complex interactions and that NaB and TSA seem to disturb this energetic equilibrium. We show for the first time that these HDACis reduce the glycolytic metabolism and increase O2 consumption coupled to ATP synthesis in H460 cells. In this scenario, the HDACis action transcend their role at the chromatin level because non-histone proteins can be acetylated and most intermediate metabolic enzymes are acetylated, including enzymes of glycolysis, fatty acid metabolism and Krebs cycle. Initially, whatever metabolic reprogramming occurred upon treatment of the cells with NaB, no gross morphological changes were observed at the level of light and electron microscopy. Likewise, the nuclear structure of treated cells was preserved, which makes it improbable that NaB had any disruptive effects on cell architecture, including intracellular compartmentation. In agreement with this view, it is worth mentioning that any known direct interaction of NaB with the cells seems to be receptor mediated, involving, for example solute transporters such as monocarboxylate transporter SMCT1. Incidentally, it has been reported that SMCT1 is usually silenced in cancer cells, a fact that may explain why relatively high concentrations of butyrate had to be used in the present work and in the literature. Indeed, TSA which is readily absorbed by the cells exerted its inhibitory effects at much lower concentrations than -Protopanaxadiol-chemical-structure.gif) NaB. Other issues relating to solute transport through the membranes of H460 cells may have a direct bearing on the results involving lactate efflux.
NaB. Other issues relating to solute transport through the membranes of H460 cells may have a direct bearing on the results involving lactate efflux.
Month: August 2019
Protective effect of knockdown of TRPM7 by RNA interference following ischemia using an in vivo model
Collectively, these studies strongly suggest that TRPM7 may be an effective pharmacological target for stroke treatment; however, compounds that could potentially be used clinically against the channel have not been identified. In this study we have Temozolomide identified the 5-LOX inhibitors NDGA, AA861, and MK886 as potent blockers of TRPM7 channel activity. The compounds were also effective at inhibiting TRPM7 channel function, as application of these molecules prevented TRPM7-induced cell rounding as well as cell death caused by low extracellular divalent cations or several forms of apoptotic stimuli. NDGA, AA861, and MK886 were originally identified by their capacity to inhibit 5-LOX, however, several lines of evidence suggest that these compounds block TRPM7 channel currents directly and independent of their inhibitory effects on 5LOX enzymatic activity. Transfection of the dsiRNA targeting 5LOX failed to lower TRPM7 whole cell currents compared to cells transfected with the control dsiRNA, although transfection of dsiRNAs targeting the 5-LOX partially interfered with TRPM7mediated cell rounding. It has been reported that 5-LOX is involved in the regulation of cell adhesion, so the effects of the 5LOX dsiRNAs on TRPM7-induced cell rounding are likely due to direct knockdown of 5-LOX expression. In addition, we were unable to reverse AA861��s blockade of TRPM7 channel activity by perfusion of the 5-LOX product 5-HPETE or its downstream metabolites into the extracellular bath solution. Likewise, inclusion of either 5-HPETE, LTD4, and LTB4 into the internal pipette solution did not prevent the inhibition of TRPM7 channel activity by AA861. Finally, the other two 5-LOX inhibitors, 5,6-DAA and zileuton, were ineffective in blocking TRPM7 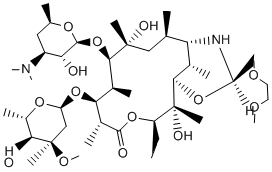 currents. Collectively, these results strongly Y-27632 dihydrochloride indicate that NDGA, AA861, and MK886 block TRPM7 channel currents independent of their actions on 5-LOX enzymatic activity. NDGA, AA861, and MK886 did not alter TRPM7 protein expression or its concentration on the cell surface, leaving it unclear how these compounds may be interfering with TRPM7 channel activity. NDGA is a lipophilic reducing agent that blocks catalysis by reducing the active site iron in 5-LOX, whereas AA861 competes with binding of arachadonic acid to the enzyme. The structurally unrelated indole-containing MK886 is also lipophilic, blocking 5-LOX activity by binding to FLAP, a membrane protein that facilitates 5-lipoxygenase enzymatic activity by enhancing the delivery of arachidonic acid to 5-LOX. Thus, the compounds may be blocking TRPM7 directly in the membrane or by interfering with binding of lipid to the channel. Since NDGA, AA861, and MK886 effectively block the endogenous TRPM7 current, a reevaluation of the results of experimental studies employing these compounds is warranted. Administration of 5-LOX inhibitors has been shown to reduce tissue damage in rodent models of cerebral ischemia and myocardial ischemia-reperfusion injury. However, no significant difference in the infarct size between control and 5-LOX knockout mice was observed using either a heart or brain model of ischemic injury. As knockdown of the TRPM7 channel reduces the pathogenesis of brain ischemia, it is tempting to speculate that 5-LOX inhibitors achieve a portion of their cellular protective effects by blocking the TRPM7 channel. Indeed, the 5-LOX inhibitors AA861 and NDGA were effective in reversing TRPM7-induced cell death when cells are cultured in low extracellular divalent cations. In addition, both knockdown of TRPM7 and application of AA861 were effective in reducing cell death caused by apoptotic stimuli.
currents. Collectively, these results strongly Y-27632 dihydrochloride indicate that NDGA, AA861, and MK886 block TRPM7 channel currents independent of their actions on 5-LOX enzymatic activity. NDGA, AA861, and MK886 did not alter TRPM7 protein expression or its concentration on the cell surface, leaving it unclear how these compounds may be interfering with TRPM7 channel activity. NDGA is a lipophilic reducing agent that blocks catalysis by reducing the active site iron in 5-LOX, whereas AA861 competes with binding of arachadonic acid to the enzyme. The structurally unrelated indole-containing MK886 is also lipophilic, blocking 5-LOX activity by binding to FLAP, a membrane protein that facilitates 5-lipoxygenase enzymatic activity by enhancing the delivery of arachidonic acid to 5-LOX. Thus, the compounds may be blocking TRPM7 directly in the membrane or by interfering with binding of lipid to the channel. Since NDGA, AA861, and MK886 effectively block the endogenous TRPM7 current, a reevaluation of the results of experimental studies employing these compounds is warranted. Administration of 5-LOX inhibitors has been shown to reduce tissue damage in rodent models of cerebral ischemia and myocardial ischemia-reperfusion injury. However, no significant difference in the infarct size between control and 5-LOX knockout mice was observed using either a heart or brain model of ischemic injury. As knockdown of the TRPM7 channel reduces the pathogenesis of brain ischemia, it is tempting to speculate that 5-LOX inhibitors achieve a portion of their cellular protective effects by blocking the TRPM7 channel. Indeed, the 5-LOX inhibitors AA861 and NDGA were effective in reversing TRPM7-induced cell death when cells are cultured in low extracellular divalent cations. In addition, both knockdown of TRPM7 and application of AA861 were effective in reducing cell death caused by apoptotic stimuli.
IDE normally regulates insulin signaling by virtue of its ability to rapidly degrade internalized pools of insulin
Members of this superfamily are commonly referred to as “inverzincins,” because they feature a zinc-binding motif that is inverted with respect to that within conventional zinc-metalloproteases. Like insulin, IDE is structurally distinctive, consisting of two bowl-shaped halves connected by a flexible linker that can switch between “open” and “closed” states. In its closed state, IDE completely encapsulates its substrates within an unusually large internal cavity that appears remarkably well-adapted to accommodate insulin. IDE degrades several other intermediate-sized peptides, including atrial natriuric peptide, glucagon, and the amyloid b-protein; however, unlike insulin, most other IDE substrates are known to be hydrolyzed by multiple proteases. Diabetes melittus is a life-threatening and highly prevalent group of endocrinological disorders that, fundamentally, are characterized by impaired insulin signaling. Correspondingly, it is the common goal of most anti-diabetic therapies to enhance insulin signaling, either by direct injection of insulin, by stimulating the production or secretion of endogenous insulin, or by activating downstream targets of the insulin receptor signaling cascade. In principle, it should be possible to enhance insulin signaling by inhibiting IDE-mediated insulin catabolism. Pharmacological inhibitors of IDE in fact attracted considerable attention in the decades following the discovery of IDE in 1949. Quite significantly, a purified PR-171 inhibitor of IDE was found to potentiate the hypoglycemic action of insulin in vivo as early as 1955. Despite more than 60 years of research on IDE and its involvement in insulin catabolism, the development of smallmolecule inhibitors of IDE has proved to be a surprisingly elusive goal. We describe herein the design, synthesis, enzymologic characterization, and enzyme-bound crystal structure of the first potent and selective inhibitors of IDE. In addition, we show that inhibition of IDE can potentiate insulin signaling within cells, by reducing the catabolism of internalized insulin. These novel IDE inhibitors represent important new pharmacological tools for the  experimental manipulation of IDE and, by extension, insulin signaling. Furthermore, our results lend new support to the old idea that pharmacological inhibition of IDE may represent an attractive approach to the treatment of diabetes mellitus. The development of Ii1 which potently inhibits IDE, but not cathepsin D enabled us for the first time to address cleanly this longstanding controversy. To that end, we conducted live-cell imaging of CHO-IR cells loaded with fluorescent insulin labeled exclusively at the Nterminus of the B chain with fluorescein isothiocyanate, a modification that has been shown not to interfere with binding to the IR. FITC-ins-loaded cells were washed then monitored for changes in PD 0332991 fluorescence in the presence of Ii1 or vehicle. In vehicle-treated cells, intracellular fluorescence decreased and extracellular fluorescence increased monotonically with time. By contrast, both intra- and extracellular fluorescence remained essentially constant in the presence of Ii1. Consistent with previous studies of insulin catabolism, the fluorescent species secreted by vehicle-treated cells were confirmed to be proteolytic fragments of FITC-ins. These results strongly suggest that the catabolism of internalized insulin is primarily, if not exclusively, carried out by IDE. Taken together, these results suggest correspondingly, insulin signaling can be potentiated significantly by inhibiting IDE proteolytic activity.
experimental manipulation of IDE and, by extension, insulin signaling. Furthermore, our results lend new support to the old idea that pharmacological inhibition of IDE may represent an attractive approach to the treatment of diabetes mellitus. The development of Ii1 which potently inhibits IDE, but not cathepsin D enabled us for the first time to address cleanly this longstanding controversy. To that end, we conducted live-cell imaging of CHO-IR cells loaded with fluorescent insulin labeled exclusively at the Nterminus of the B chain with fluorescein isothiocyanate, a modification that has been shown not to interfere with binding to the IR. FITC-ins-loaded cells were washed then monitored for changes in PD 0332991 fluorescence in the presence of Ii1 or vehicle. In vehicle-treated cells, intracellular fluorescence decreased and extracellular fluorescence increased monotonically with time. By contrast, both intra- and extracellular fluorescence remained essentially constant in the presence of Ii1. Consistent with previous studies of insulin catabolism, the fluorescent species secreted by vehicle-treated cells were confirmed to be proteolytic fragments of FITC-ins. These results strongly suggest that the catabolism of internalized insulin is primarily, if not exclusively, carried out by IDE. Taken together, these results suggest correspondingly, insulin signaling can be potentiated significantly by inhibiting IDE proteolytic activity.
To gain further insight on the therapeutic potential of these iodinated TTR fibrillogenesis inhibitors in vitro binding
Recent studies on the aggregation pathway of TTR into amyloid fibrils point to a fibrillogenesis model which involves several steps such as dissociation of the tetramer, changes on monomer conformation, aggregation of conformationally modified monomers into non-fibrillar oligomers that latter form protofibrils and further elongate into mature fibrils. This mechanism along with the fact that binding of thyroid hormones to TTR results in tetramer stabilization, suggests that inhibition of amyloid fibril formation can be accomplished by small molecule compounds sharing structural similarities with T4. Indeed this Rapamycin hypothesis has been confirmed by the identification of several families of compounds that, by binding to TTR, stabilize the ground state of the protein to an extent which is proportional to the dissociation constants. The most common molecular features on this range of inhibitors is that they are composed of two aromatic rings bearing halogen substituents in one moiety and hydrophilic functions in the second which give rise to structures as diverse as tetrahydroquinolines, dihydropyridines, benzodiazepines, phenoxazines, stilbenes and benzoxazoles. Thyroid hormones are the only human biochemicals presenting multiple iodine atoms in their molecules. Blake and co-workers were the first to describe that in each TTR binding site there are six pockets capable of accomodate an iodine atom. Indeed, when T4 binds TTR, four of these six pockets become occupied by the iodine atoms of the hormone molecule resulting in a close steric fit between the ligand and the binding site. Therefore, iodine atoms are crucial for the binding mode of thyroid hormones to TTR, making an important contribution to the protein-hormone interactions that stabilise the complex. In spite of this evidence, up to our knowledge, none of the potential newly designed TTR amyloid inhibitors have taken advantage of the potential benefits of incorporating iodine atoms to mimick the iodine-assisted binding mode of thyroid hormones. Accordingly, the aim of the present investigation was to provide initial evidences for the hypothesis that iodine atom addition to already known TTR inhibitors could produce more potent TTR fibrillogenesis inhibitors. Salicylates look particularly interesting as drug candidates due to their long therapeutic tradition and wide clinical applications. Owing that a number of salicylate analogues have also been postulated as good TTR amyloid inhibitors and because the salicylic core is RWJ 64809 amenable to electrophilic iodination, a salicylate was chosen as a model template to test this hypothesis. The positioning of iododiflunisal in the TTR channel is exclusively in the forward mode, this is, with the difluorophenyl ring occupying the inner part of the cavity and the salicylic ring the outer part. This is a common feature among other inhibitors having a biphenyl core molecule. The same forward mode is also the single disposition that is seen in both 23b and 22b structures which show almost coincident spatial ring disposition. In both cases, the compounds are located further inside in the cavity than iododiflunisal. In sharp contrast, diflunisal is observed in the pocket sharing two orientations with equal probabilities, the one described as forward and a totally opposite where the rings swap positions that is called reverse mode. The iodine atom in the iododiflunisal complex establishes close hydrophobic interactions with Leu17, Thr106, Ala108, Thr119 and Val121, thus, occupying the HBP1 pocket which is the outermost and more hydrophobic HBP. The innermost HBP pockets, HBP3 and HBP39, in turn, closely interact with the fluorine atoms of the difluorophenyl ring. A further stabilizing interaction is found between the carbonyl group of Thr106 and iodine which closely resembles an halogen bond. Similar but more optimized interactions than in  the iododiflunisal complex are observed for the iodine atom in both crystal structures of 23b and 22b complexes.
the iododiflunisal complex are observed for the iodine atom in both crystal structures of 23b and 22b complexes.
In the latter oseltamivir and zanamivir are widely used against influenza effectively reducing the duration
The former was developed earlier and most influenza viruses presently circulating among humans are resistant against the inhibitors from this group. These drugs were the only available options during the 2009 pandemic. Influenza type A and B viruses contain three major surface proteins, HA, NA and M2. HA mediates viral attachment to host cells by binding sialic acids on carbohydrate side chains of cell surface glycoproteins and glycolipids. HA also mediates virus entry into host cells through the fusion of the viral envelope with the  endosomal membrane. As fusion occurs, the viral genome is released into cytoplasm of host cells by the aid of the M2 protein ion channel. NA cleaves the terminal sialic acid residues on oligosaccharide chains that serve as viral HA receptors. Through this enzymatic activity, NA plays an important role in the spread of infection from cell to cell because virions stick to the cell surface or aggregate with each other if sialic acid residues are not removed from the surface of infected cells and progeny viruses. In body fluids, numerous molecules containing sialic acid exist and most of them are able to bind to virus HA and inhibit the hemagglutination activity of influenza virus. Human saliva has been reported to contain such hemagglutination inhibitors. During the initial infection of mucosal epithelial cells, influenza virus encounters these inhibitory molecules in mucus and viral NA is speculated to inactivate such inhibitors so that viral HA is able to bind to receptors on the surface of epithelial cells. Influenza virus initiates infection in the upper respiratory tract where commensal bacterial flora exists. The synergism between influenza virus and bacteria has been documented in past influenza outbreaks. It was first observed when the swine influenza virus was discovered by Shope in 1931. He indicated that the isolated virus and Haemophilus influenzae acted together to produce swine influenza and that neither alone was capable of inducing disease. Furthermore, reexamination of samples from the influenza MG132 Proteasome inhibitor pandemic of 1918 indicated that the majority of Nilotinib abmole patients died of secondary bacterial pneumonia. In the influenza pandemic of 1957-1958, most deaths attributed to influenza A virus infection occurred concurrently with bacterial pneumonia. Moreover, recent postmortem studies among fatal Apdm09 cases from the 2009 pandemic established a link between bacterial lung infections and increased mortality or developing complications. Mechanisms for the synergy between bacteria and influenza viruses involve the activity of either bacterial or viral enzymes. For influenza virus to obtain membrane fusion activity, HA protein has to be cleaved by a host proteinase. Some strains of Staphylococcus aureus secrete a protease which significantly influences the outcome of influenza infection by cleavage activation of HA. Influenza virus NA, on the other hand, potentiates the development of pneumonia by stripping sialic acid from lung cells, thus exposing receptors for Streptococcus pneumoniae adhesion. Classical studies on influenza virus receptors by Gottschalk showed that neuraminidase treatment inactivates hemagglutination inhibitors in serum and mucus secretions by removing the sialic acid residues of oligosaccharide chains on the inhibitors. The most well-known source of neuraminidase used for this purpose is a so-called receptor-destroying enzyme. It has been shown by several groups that influenza A viruses lacking neuraminidase activity can undergo multiple cycles of replication in an in vitro infection system if bacterial neuraminidase is provided exogenously.
endosomal membrane. As fusion occurs, the viral genome is released into cytoplasm of host cells by the aid of the M2 protein ion channel. NA cleaves the terminal sialic acid residues on oligosaccharide chains that serve as viral HA receptors. Through this enzymatic activity, NA plays an important role in the spread of infection from cell to cell because virions stick to the cell surface or aggregate with each other if sialic acid residues are not removed from the surface of infected cells and progeny viruses. In body fluids, numerous molecules containing sialic acid exist and most of them are able to bind to virus HA and inhibit the hemagglutination activity of influenza virus. Human saliva has been reported to contain such hemagglutination inhibitors. During the initial infection of mucosal epithelial cells, influenza virus encounters these inhibitory molecules in mucus and viral NA is speculated to inactivate such inhibitors so that viral HA is able to bind to receptors on the surface of epithelial cells. Influenza virus initiates infection in the upper respiratory tract where commensal bacterial flora exists. The synergism between influenza virus and bacteria has been documented in past influenza outbreaks. It was first observed when the swine influenza virus was discovered by Shope in 1931. He indicated that the isolated virus and Haemophilus influenzae acted together to produce swine influenza and that neither alone was capable of inducing disease. Furthermore, reexamination of samples from the influenza MG132 Proteasome inhibitor pandemic of 1918 indicated that the majority of Nilotinib abmole patients died of secondary bacterial pneumonia. In the influenza pandemic of 1957-1958, most deaths attributed to influenza A virus infection occurred concurrently with bacterial pneumonia. Moreover, recent postmortem studies among fatal Apdm09 cases from the 2009 pandemic established a link between bacterial lung infections and increased mortality or developing complications. Mechanisms for the synergy between bacteria and influenza viruses involve the activity of either bacterial or viral enzymes. For influenza virus to obtain membrane fusion activity, HA protein has to be cleaved by a host proteinase. Some strains of Staphylococcus aureus secrete a protease which significantly influences the outcome of influenza infection by cleavage activation of HA. Influenza virus NA, on the other hand, potentiates the development of pneumonia by stripping sialic acid from lung cells, thus exposing receptors for Streptococcus pneumoniae adhesion. Classical studies on influenza virus receptors by Gottschalk showed that neuraminidase treatment inactivates hemagglutination inhibitors in serum and mucus secretions by removing the sialic acid residues of oligosaccharide chains on the inhibitors. The most well-known source of neuraminidase used for this purpose is a so-called receptor-destroying enzyme. It has been shown by several groups that influenza A viruses lacking neuraminidase activity can undergo multiple cycles of replication in an in vitro infection system if bacterial neuraminidase is provided exogenously.