Furthermore, examination of changes in the phosphoproteome of rape seeds during the filling stage identified 70 phosphoproteins and 16 non-redundant phosphoproteins, which were verified by mapping the phosphorylation sites. Analysis of vacuolar and cell membranes of rice bud and root revealed 230 membrane and membrane-associated proteins, 20% of which are phosphorylated. In addition, soybean root hairs contain 1625 unique phosphopeptides, including 1659 nonredundant phosphorylation sites, which originate from 1126 phosphoproteins. A recent study identified multiple components of ABA-responsive protein phosphorylation network by integrating genetics with phosphoproteomics. Furthermore, Wang et al. have shown the role of the SnRK2 protein kinases in the ABA signaling pathway by using quantitative phosphopro teomics. To date, only a few plant species have been annotated with protein phosphorylation data and these do not include the economically important plant, cotton. We previously examined the proteome of cotton leaves in response to NO treatment. To follow-up this research, we performed quantitative time-course measurements of the phosphoproteome of cotton leaves treated with sodium nitroprusside, a NO donor. This is the first study to show that NO exposure in leaf tissue alters the phosphorylation states of multiple proteins found in cotton. This information should accelerate research on NO metabolic regulation and will lay a novel, theoretical foundation for further related studies in cotton. The neurotrophins are classically known for their participation in the Staurosporine development, growth, function, and survival of neurons in both the central and peripheral GSI-IX nervous system. Although abundant in the nervous system, BDNF and Ntrk2 are expressed in other cell types and tissues, and BDNF mRNA is found in the majority of the human body organs. In humans, mature BDNF is sequestered in platelets and released upon their degranulation. As such, BDNF has access to all tissues and organs. Motile cells including activated T cells, B cells, and monocytes have been shown to express BDNF in vitro, as have eosinophils, dendritic cells, and endothelial cells. In mice, the visceral epithelium, and airway epithelium are significant sources of BDNF. As for Ntrk2, a comprehensive analysis of Ntrk2 immunoreactivity was assessed and it was found to be expressed mainly in glandular cells of the salivary gland, small intestine, colon, endocrine pancreas, bone marrow hematopoietic cells, monocytes/macrophages of the lymph nodes and spleen, and in the epidermis. Previous studies have shown  that neurotrophins in the brain are regulated by neuronal activity, and steroid hormones, and that tissue-specific expression is driven by multiple promoters. While BDNF and Ntrk2 expression has been demonstrated in some reproductive tissues including the ovary, and placenta, their uterine expression under physiological conditions has been questionable. BDNF expression was demonstrated by immunohistochemistry in the mouse, and human uterus and by in situ hybridization in the mouse, and rat uterus. While Ntrk2 could not be detected in the mouse and human uterus, others have been successful. To date only one study has looked for the presence of both ligand and receptor simultaneously, in the murine uterus.
that neurotrophins in the brain are regulated by neuronal activity, and steroid hormones, and that tissue-specific expression is driven by multiple promoters. While BDNF and Ntrk2 expression has been demonstrated in some reproductive tissues including the ovary, and placenta, their uterine expression under physiological conditions has been questionable. BDNF expression was demonstrated by immunohistochemistry in the mouse, and human uterus and by in situ hybridization in the mouse, and rat uterus. While Ntrk2 could not be detected in the mouse and human uterus, others have been successful. To date only one study has looked for the presence of both ligand and receptor simultaneously, in the murine uterus.
Month: August 2019
In computational modeling of cellulose protofibril formation simulated the interactions
For further understanding of FF-TEM views of SP600125 cellular membranes and standard FF-TEM nomenclature, see. Clarifying some aspects of FF-TEM imaging will aid the understanding of our results. When shadowing metal is applied from a fixed angle in FF-TEM, the replica appears three dimensional due to the differential interaction of electron dense metal with the changing topography of the sample surface. The shadowing metal accumulates on a raised intramembrane protein or cellular surface on the side proximal to the shadowing source. Relatively tall objects block the deposition of metal on the side opposite to the shadowing source resulting in electrontranslucent, white, ‘shadows’. If an object is nearly flat relative to the overall sample surface it may be almost uniformly highlighted by a thin metal coating that does not obscure it, as seen below for rosettes CSCs on the ES of the Reversine customer reviews plasma membrane. In differentiating TEs, the patterned secondary wall thickenings push down into the cytoplasm and cause undulations in  the topography of the plasma membrane. In the lower area of the TE, ‘sw’ marks a plasma membrane trough where the secondary cell wall was completely removed by the fracture process to reveal the plasma membrane ES. In the middle region of the TE, ‘sw’ marks a secondary wall thickening where cell wall material still fills the trough on the left side. A few fibrils, putatively cellulose, remain on the ES of the plasma membrane, and their average diameter was 4.15 nm after subtraction of two times the approximate thickness of the shadowing metal. Sometimes the fibrils appeared irregular on the edges as would be consistent with recent synthesis and nascent crystallization near the extrusion point. Some of the fibrils have hemispherical ends. Other hemispheres are in the vicinity of fibrils, but are not clearly contiguous with one fibril. The average diameter of the hemispheres was 23.9 nm, which is much larger than the typical IMPs scattered throughout the membrane. The hemispheres and the fibrils are shadowed objects that lie on top of the plasma membrane bilayer, but in the same plane. Consistently, all of the images show that the rosette CSCs are nearly flat in the membrane because the six individual globules did not cast shadows. In contrast, some other unidentified IMPs projected higher above the membrane surface and cast white shadows. These observations were consistent with images from other TEs. Often the plasma membrane bilayer splits during fracture and rosette CSCs partition with the protoplasmic fracture face of the plasma membrane as shown in numerous past studies and our work as well. Correspondingly, we sometimes saw holes in the extracellular leaflet of the plasma membrane where the TMHs of CESA had pulled out as the plasma membrane split during fracture. Figure 8B shows another view of a rosette CSC in a small depression of the plasma membrane ES. Figure 8C shows a rosette CSC in the plasma membrane ES in close proximity to a curving fibril about 4 nm wide. Numerous longer fibrils along with a rosette CSC in the plasma membrane ES are shown in Figure 8D. Two fibrils with globular structures at opposite ends are shown in Figure 8E, an observation that is consistent with the bidirectional travel of CESAs that was demonstrated by live-cell imaging during synthesis of epidermal cell walls in hypocotyls.
the topography of the plasma membrane. In the lower area of the TE, ‘sw’ marks a plasma membrane trough where the secondary cell wall was completely removed by the fracture process to reveal the plasma membrane ES. In the middle region of the TE, ‘sw’ marks a secondary wall thickening where cell wall material still fills the trough on the left side. A few fibrils, putatively cellulose, remain on the ES of the plasma membrane, and their average diameter was 4.15 nm after subtraction of two times the approximate thickness of the shadowing metal. Sometimes the fibrils appeared irregular on the edges as would be consistent with recent synthesis and nascent crystallization near the extrusion point. Some of the fibrils have hemispherical ends. Other hemispheres are in the vicinity of fibrils, but are not clearly contiguous with one fibril. The average diameter of the hemispheres was 23.9 nm, which is much larger than the typical IMPs scattered throughout the membrane. The hemispheres and the fibrils are shadowed objects that lie on top of the plasma membrane bilayer, but in the same plane. Consistently, all of the images show that the rosette CSCs are nearly flat in the membrane because the six individual globules did not cast shadows. In contrast, some other unidentified IMPs projected higher above the membrane surface and cast white shadows. These observations were consistent with images from other TEs. Often the plasma membrane bilayer splits during fracture and rosette CSCs partition with the protoplasmic fracture face of the plasma membrane as shown in numerous past studies and our work as well. Correspondingly, we sometimes saw holes in the extracellular leaflet of the plasma membrane where the TMHs of CESA had pulled out as the plasma membrane split during fracture. Figure 8B shows another view of a rosette CSC in a small depression of the plasma membrane ES. Figure 8C shows a rosette CSC in the plasma membrane ES in close proximity to a curving fibril about 4 nm wide. Numerous longer fibrils along with a rosette CSC in the plasma membrane ES are shown in Figure 8D. Two fibrils with globular structures at opposite ends are shown in Figure 8E, an observation that is consistent with the bidirectional travel of CESAs that was demonstrated by live-cell imaging during synthesis of epidermal cell walls in hypocotyls.
Efflux a variety of structurally and functionally diverse chemotherapeutic drugs out of cancer cells
Thereby reducing the intracellular drug accumulation, increasing the likelihood of decreased cytotoxic and thus unsuccessful treatment. Currently, 48 distinct ABC transporters have been identified in the human genome, and these can further divided into seven subfamilies based on sequence similarities. Among these transporters, the ABCB1 transporter is the most important mediator of MDR, and is responsible for chemotherapeutic drug resistance to a variety of drug, including vinca alkaloids, anthracyclines, epipodophyllotoxins and taxanes. The overexpression of ABCB1 occurs in 40– 50% of cancer patients, and is associated with a poor clinical outcome. Based on these findings, a number of studies have attempted to selectively inhibit ABCB1 activity as a strategy to reverse MDR in cancer chemotherapy. Indeed, in the past 30 years, significant efforts have been made to design and test specific ABCB1 inhibitors and this has resulted in the development of three generations of ABCB1 inhibitors. CX-4945 However, currently, none of the compounds in the three generations have been approved for clinical use. The first-generation ABCB1 inhibitors, including verapamil, quinine, and cyclosporin A lacked selectivity and produced undesirable adverse effects at plasma concentrations necessary to inhibit ABCB1. The second-generation ABCB1 inhibitors, such as valspodar/PSC-833 and biricodar/VX-710, had improved tolerability compared to the first-generation compounds. However, they produced unpredictable interactions with other transport proteins and inhibited CYP3A4, one of the major chemotherapeutic drug metabolizing enzymes, thereby reducing the the clearance and metabolism of chemotherapeutic drugs. The third-generation inhibitors were more selective for the ABCB1 transporters in ongoing clinical trials. Nonetheless, some of these compounds produced significant adverse effects and had an unfavorable pharmacokinetic profile, including poor solubility as well as reducing the clearance of clinically used anticancer drugs. Recent results from our laboratory and others indicate that several tyrosine kinase inhibitors, including imatinib, nilotinib, lapatinib, and erlotinib, can reverse MDR to antineoplastic drugs mediated by ABCtransporters. However, the reversal potential of these TKIs have not been determined in clinical trials. Consequently, it is necessary to develop more efficacious, non-toxic and less expensive compounds to reverse MDR in cancer cells. In the course of our search for compounds that reverse MDR, we found that vardenafil and tadalafil, two phosphodiesterase PF-04217903 type-5 inhibitors clinically used in the treatment of male erectile dysfunction, significantly reversed ABCB1-mediated MDR. In the present study, we conducted experiments to ascertain the reversal mechanism of vardenafil and tadalafil in ABCB1 overexpressing cancer cells. In addition, we also examined their effect on other major ABC drug transporters such as MRP1 and BCRP. One of the major mechanisms responsible to MDR in cancer cells is the overexpression of the ABCB1 transporter. However, currently, none of the ABCB1 inhibitors or modulators have been approved for clinical oncological practice. The present study demonstrates for the first time that vardenifil, a PDE-5 inhibitor used in the treatment of male erectile dysfunction, reverses ABCB1-mediated MDR in a  concentration-dependent manner.
concentration-dependent manner.
Considered as additional targets in the synergistic cooperation with proteasome inhibitors
Since they all are inhibited by cantharidin, and have been implicated in contributing to apoptosis regulation. The synthesis of a salubrinal-derived affinity reagent may therefore be critical to pinpoint the exact molecular target of this inhibitor and to assist in shedding further light on its mode of action. Identification of the phosphatase targeted by salubrinal  will also help to identify the corresponding phosphatase substrates and signaling pathways that are participating in survival regulation. Proteasome inhibitors exert considerable cytostatic and cytotoxic effects in particular cancer cells types already as single ICI 182780 agents, but they may be even more useful as sensitizers to apoptosis induction when delivered in combination with other anticancer drugs. Given the synergistic enhancement of proteasome inhibitor toxicity by salubrinal in K562 and other leukemic cells, salubrinal may therefore very well be added to the growing list of drugs that cooperate with proteasome inhibitor to kill hemopoietic tumor cells. It may be speculated that cancer patients receiving proteasome inhibitor treatment could benefit from the coadministration of salubrinal also for a second reason: While enhancing the killing of sensitized leukemic cells, salubrinal may at the same time ameloriate proteasome inhibitor-mediated toxicity in neuronal cells, Saveguarding neuronal cells by this means would be a desirable feature e.g. for myeloma patients receiving proteasome inhibitor treatment, since development of peripheral neuropathy is one of the major side effects and could be a direct consequence of the impairment of the ubiquitin-proteasome system. Further investigations will reveal, whether salubrinal or derivatives thereoff can be included in a therapeutic strategy that is based on the induction of ER stress and maintains a strong and selective toxicity for the tumor cells on the one hand but confers protection to neuronal and other non-transformed cells on the other. These studies will have to consider also the possibility that salubrinal may exert other side effects, due to the pleiotropic nature of phosphatase inhibitors. However, a recent proteomic study demonstrated that the number of proteins actually affected by salubrinal treatment appeared to be very limited, suggesting that salubrinal may R428 possess unique features that renders it interesting enough to further develop it into a clinically useful compound. The data presented here in summary support a paradigm shift on the protective role of the phosphatase inhibitor salubrinal during ER stress, as this compound can obviously also augment apoptosis, depending on the specific ER-stress signal and the cellular system investigated. They also suggest that the concomitant targeting of specific phosphatases in a proteasome inhibitorbased strategy to kill cancer cells could be an attractive option. The resistance of tumor cells to a variety of structurally and mechanistically unrelated cytotoxic drugs, also known as multidrug resistance, is one of the major obstacles in the successful treatment of cancer. It is estimated that approximately 500,000 new cases of cancer each year exhibit the drug resistant phenotype. One of the known causes of MDR is overexpression of the ATP-binding cassette transporters, such as P-glycoprotein, multidrug resistance proteins and breast cancer resistant protein.
will also help to identify the corresponding phosphatase substrates and signaling pathways that are participating in survival regulation. Proteasome inhibitors exert considerable cytostatic and cytotoxic effects in particular cancer cells types already as single ICI 182780 agents, but they may be even more useful as sensitizers to apoptosis induction when delivered in combination with other anticancer drugs. Given the synergistic enhancement of proteasome inhibitor toxicity by salubrinal in K562 and other leukemic cells, salubrinal may therefore very well be added to the growing list of drugs that cooperate with proteasome inhibitor to kill hemopoietic tumor cells. It may be speculated that cancer patients receiving proteasome inhibitor treatment could benefit from the coadministration of salubrinal also for a second reason: While enhancing the killing of sensitized leukemic cells, salubrinal may at the same time ameloriate proteasome inhibitor-mediated toxicity in neuronal cells, Saveguarding neuronal cells by this means would be a desirable feature e.g. for myeloma patients receiving proteasome inhibitor treatment, since development of peripheral neuropathy is one of the major side effects and could be a direct consequence of the impairment of the ubiquitin-proteasome system. Further investigations will reveal, whether salubrinal or derivatives thereoff can be included in a therapeutic strategy that is based on the induction of ER stress and maintains a strong and selective toxicity for the tumor cells on the one hand but confers protection to neuronal and other non-transformed cells on the other. These studies will have to consider also the possibility that salubrinal may exert other side effects, due to the pleiotropic nature of phosphatase inhibitors. However, a recent proteomic study demonstrated that the number of proteins actually affected by salubrinal treatment appeared to be very limited, suggesting that salubrinal may R428 possess unique features that renders it interesting enough to further develop it into a clinically useful compound. The data presented here in summary support a paradigm shift on the protective role of the phosphatase inhibitor salubrinal during ER stress, as this compound can obviously also augment apoptosis, depending on the specific ER-stress signal and the cellular system investigated. They also suggest that the concomitant targeting of specific phosphatases in a proteasome inhibitorbased strategy to kill cancer cells could be an attractive option. The resistance of tumor cells to a variety of structurally and mechanistically unrelated cytotoxic drugs, also known as multidrug resistance, is one of the major obstacles in the successful treatment of cancer. It is estimated that approximately 500,000 new cases of cancer each year exhibit the drug resistant phenotype. One of the known causes of MDR is overexpression of the ATP-binding cassette transporters, such as P-glycoprotein, multidrug resistance proteins and breast cancer resistant protein.
We combined in silico pharmacophore modeling with subsequent in vitro assays to systematically investigate
Which are FDA-approved agents, and 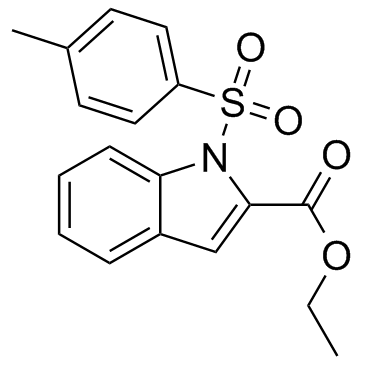 the non-FDA-labeled PPI tenatoprazole. The pharmacophore models described for OCT1 and OCT2 share a hydrophobic interaction site and a positive ionizable site. The pharmacophore models of the present study are in line with these models in having at least 1 hydrophobic interaction site as well. The lack of a positive ionizable site in our models is probably due to the fact that many of the compounds selected for the training sets are neutral at pH 7.4. Our pharmacophore models predict PPIs to be very potent inhibitors of OCT1, OCT2, and OCT3, mainly due to their hydrophobic features and presence of H-bond acceptor sites. In order to validate the data of the in silico pharmacophore modeling, we generated cell systems stably expressing recombinant human OCT1, OCT2, or OCT3. All 3 transfected HEK cell lines BIBW2992 expressed functionally active organic cation transporters as demonstrated by time-dependent TEA and metformin uptake, which are both well-established substrates of OCTs. Consistent with these functional data, the recombinant OCT proteins were detected in the plasma membrane of the OCT-expressing HEK cells as well as in membrane fractions from these cells as expected. The most striking result of our study was a potent inhibition of metformin uptake transport by all five PPIs for all 3 OCT proteins tested with IC50 values in the low micromolar range, similar to calculated total PPI concentrations in portal venous blood. Moreover, we could clearly show that none of these PPIs are substrates for the 3 OCT transport proteins. The fact that drugs are potent OCT inhibitors without being substrates, is in agreement with results obtained for several other compounds. Moreover, OCT1- and OCT3-mediated metformin uptake appears to be activated by low concentrations of selected PPIs, which is in line with previous observations reported for carvedilol and OCT2-mediated metformin uptake but also for other uptake transporters and inhibitors. However, underlying molecular mechanisms are currently unknown. Given the role of OCT1 for metformin action and of OCT2 for renal secretion of metformin, efforts have been made to identify physicochemical parameters that may predict whether a compound inhibits the OCT transporters. One study showed that a positive charge at pH 7.4 and a high lipophilicity are the main properties of potent OCT1 inhibitors. The PLS analysis revealed that the ClogP value likewise appears to be a relevant factor for Rapamycin explaining OCT1 inhibition by the 5 PPIs. For OCT2, one study also identified the ClogP value as a principal factor for potent inhibition, while in another study the TPSA value was predictive for inhibition. However, neither the ClogP value nor the TPSA value are apparently predictive for OCT2 or OCT3 inhibition by PPIs. It therefore remains unclear which physicochemical parameters determine the inhibition potency of PPIs towards OCT2 and OCT3. Another physicochemical parameter, i.e. the charge at pH 7.4 that was identified as a relevant property of OCT1 inhibitors, is apparently not sufficient for predicting a compound��s inhibition potencytowardsOCTs since PPIs are neutral at pH 7.4 and it has been shown that several other OCT inhibitors are likewise not positively charged. Currently, to the best of our knowledge no interaction studies in healthy volunteers and/or patients exist elucidating pharmacokinetic and/or �Cdynamic consequences of a combined therapy of metformin and PPIs.
the non-FDA-labeled PPI tenatoprazole. The pharmacophore models described for OCT1 and OCT2 share a hydrophobic interaction site and a positive ionizable site. The pharmacophore models of the present study are in line with these models in having at least 1 hydrophobic interaction site as well. The lack of a positive ionizable site in our models is probably due to the fact that many of the compounds selected for the training sets are neutral at pH 7.4. Our pharmacophore models predict PPIs to be very potent inhibitors of OCT1, OCT2, and OCT3, mainly due to their hydrophobic features and presence of H-bond acceptor sites. In order to validate the data of the in silico pharmacophore modeling, we generated cell systems stably expressing recombinant human OCT1, OCT2, or OCT3. All 3 transfected HEK cell lines BIBW2992 expressed functionally active organic cation transporters as demonstrated by time-dependent TEA and metformin uptake, which are both well-established substrates of OCTs. Consistent with these functional data, the recombinant OCT proteins were detected in the plasma membrane of the OCT-expressing HEK cells as well as in membrane fractions from these cells as expected. The most striking result of our study was a potent inhibition of metformin uptake transport by all five PPIs for all 3 OCT proteins tested with IC50 values in the low micromolar range, similar to calculated total PPI concentrations in portal venous blood. Moreover, we could clearly show that none of these PPIs are substrates for the 3 OCT transport proteins. The fact that drugs are potent OCT inhibitors without being substrates, is in agreement with results obtained for several other compounds. Moreover, OCT1- and OCT3-mediated metformin uptake appears to be activated by low concentrations of selected PPIs, which is in line with previous observations reported for carvedilol and OCT2-mediated metformin uptake but also for other uptake transporters and inhibitors. However, underlying molecular mechanisms are currently unknown. Given the role of OCT1 for metformin action and of OCT2 for renal secretion of metformin, efforts have been made to identify physicochemical parameters that may predict whether a compound inhibits the OCT transporters. One study showed that a positive charge at pH 7.4 and a high lipophilicity are the main properties of potent OCT1 inhibitors. The PLS analysis revealed that the ClogP value likewise appears to be a relevant factor for Rapamycin explaining OCT1 inhibition by the 5 PPIs. For OCT2, one study also identified the ClogP value as a principal factor for potent inhibition, while in another study the TPSA value was predictive for inhibition. However, neither the ClogP value nor the TPSA value are apparently predictive for OCT2 or OCT3 inhibition by PPIs. It therefore remains unclear which physicochemical parameters determine the inhibition potency of PPIs towards OCT2 and OCT3. Another physicochemical parameter, i.e. the charge at pH 7.4 that was identified as a relevant property of OCT1 inhibitors, is apparently not sufficient for predicting a compound��s inhibition potencytowardsOCTs since PPIs are neutral at pH 7.4 and it has been shown that several other OCT inhibitors are likewise not positively charged. Currently, to the best of our knowledge no interaction studies in healthy volunteers and/or patients exist elucidating pharmacokinetic and/or �Cdynamic consequences of a combined therapy of metformin and PPIs.