Increasing evidence suggests that some proteasome inhibitors exibit an allosteric effect on proteasome stability; MG262 treated purified 26S proteasomes were resistant to apyrase-induced proteasome dissociation whereas MG132 had no effect on proteasome stability. In other studies, bortezomib was reported to activate the beta 2 subunit, which cleaves at basic amino acids. The previous peptidomic study with epoxomicin noted that many of the peptides which were elevated by this compound contained an acidic residue in the P1 position of the cleavage site required to produce these peptides. Because epoxomicin does not inhibit the beta 1 AG-013736 319460-85-0 subunit responsible for cleavage at acidic residues, it would be expected that inhibition of the beta 2 and beta 5 subunits would lead to a greater share of protein degradation occurring at acidic residues. However, some of the peptides that were elevated upon treatment of cells with epoxomicin, and most of the peptides elevated upon treatment of cells with bortezomib, have hydrophobic residues in the P1 position of the cleavage site. Similarly, carfilzomib and MG262 also elevated levels of peptides that required cleavage at hydrophobic sites; all of these inhibitors are most potent at the beta 5 subunit, which is responsible for cleaving at hydrophobic sites. Somehow the inhibitors of the beta 5 subunit appear to be activating the beta 5 subunit, possibly by affecting the opening of the gate within the 20S proteasome core particle; bortezomib, MG262, and epoxomicin were all found to open this 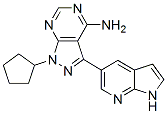 gate. In the present study, we found that bortezomib showed EX 527 comparable inhibition of the 20S core particle and an open-gate mutant of this 20S core particle when assayed with the standard substrate for beta 5 activity, but it is possible that allosteric regulation of the proteasome affects the intracellular peptides differently than the synthetic substrate. For example, Kisselev et al found that hydrophobic peptides including SuccLeu-Leu-Val-Tyr-AMC can trigger gate opening and stimulate the activity of 20S particles. A related possibility is that the various proteasome forms are differentially affected by inhibitors. In support of this hypothesis, the antiviral drug ritonavir was found to activate the chymotryptic-like activity of the 26S form of the proteasome while inhibiting the 20S form. Although we found no difference in the effect of bortezomib on the chymotryptic-like activity of the 26S versus the 20S form, or the 20S form activated by Blm10, it remains possible that allosteric effects of the proteasome inhibitors influence cleavage of the intracellular peptides by the various proteasome forms. Our results do not support the hypothesis that the proteasome inhibitors have off-target effects on enzymes that further degrade the peptides produced by the proteasome. A number of different enzymes have been implicated in the degradation of proteasome products, including oligopeptidases, aminopeptidases, and TPP2. Based on a bioinformatic approach, it was proposed that bortezomib could be an inhibitor of TPP2, although no direct evidence of this was provided. In the present study, we could not detect any inhibition of TPP2 activity by bortezomib. Furthermore, peptidomic analysis of cells treated with butabindide, a potent and selective TPP2 inhibitor, did not produce dramatic changes in the cellular peptidome. These results suggest that the bortezomib-induced changes in the cellular peptidome are not due to inhibition of TPP2. The failure of butabindide to cause large changes in the cellular peptidome suggests that TPP2 does not play a major role in the degradation of the intracellular peptides detected with the peptidomic technique, consistent with the finding that TPP2 is not required for the production of peptides bound to HLA.
gate. In the present study, we found that bortezomib showed EX 527 comparable inhibition of the 20S core particle and an open-gate mutant of this 20S core particle when assayed with the standard substrate for beta 5 activity, but it is possible that allosteric regulation of the proteasome affects the intracellular peptides differently than the synthetic substrate. For example, Kisselev et al found that hydrophobic peptides including SuccLeu-Leu-Val-Tyr-AMC can trigger gate opening and stimulate the activity of 20S particles. A related possibility is that the various proteasome forms are differentially affected by inhibitors. In support of this hypothesis, the antiviral drug ritonavir was found to activate the chymotryptic-like activity of the 26S form of the proteasome while inhibiting the 20S form. Although we found no difference in the effect of bortezomib on the chymotryptic-like activity of the 26S versus the 20S form, or the 20S form activated by Blm10, it remains possible that allosteric effects of the proteasome inhibitors influence cleavage of the intracellular peptides by the various proteasome forms. Our results do not support the hypothesis that the proteasome inhibitors have off-target effects on enzymes that further degrade the peptides produced by the proteasome. A number of different enzymes have been implicated in the degradation of proteasome products, including oligopeptidases, aminopeptidases, and TPP2. Based on a bioinformatic approach, it was proposed that bortezomib could be an inhibitor of TPP2, although no direct evidence of this was provided. In the present study, we could not detect any inhibition of TPP2 activity by bortezomib. Furthermore, peptidomic analysis of cells treated with butabindide, a potent and selective TPP2 inhibitor, did not produce dramatic changes in the cellular peptidome. These results suggest that the bortezomib-induced changes in the cellular peptidome are not due to inhibition of TPP2. The failure of butabindide to cause large changes in the cellular peptidome suggests that TPP2 does not play a major role in the degradation of the intracellular peptides detected with the peptidomic technique, consistent with the finding that TPP2 is not required for the production of peptides bound to HLA.