Signifying that some AhR activity is necessary to keep inflammatory protein levels under control. It would not be unreasonable to assume that endogenous AhR ligands present in organs such as the lung maintain constitutive AhR activity at levels that do not cause alterations in gene expression but are sufficient to prevent an exaggerated inflammatory response. Such selective modulation of AhR activity could be why activation of the AhR by CSE repressed COX-2 expression, whereas classic AhR ligands such as TCDD increase COX-2 protein levels. Both CSE and BP increased AhR activation in lung fibroblasts, as evaluated by Cyp1a1 mRNA induction, suggesting that the incongruity of results between CSE and BP is not due to inability of CSE to activate the AhR. Discrepancy in physiological responses to AhR ligands have been observed elsewhere, including murine models of multiple sclerosis. Here, activation of the AhR by either TCDD or ITE suppresses experimental autoimmune encephalomyelitis yet is enhanced by FICZ. Moreover, both ITE and TCDD elicit the same pattern of AhR-dependent gene expression, yet ITE does not cause toxicological outcomes associated with dioxin exposure, suggesting that their divergent mechanisms of action may be independent of classic AhR activation. The AhR binds to a structurally-diverse array of ligands, and it has been postulated that differential binding to the AhR may contribute to divergence in overall functionality. The high plasticity of ligand effects on signaling pathways, including ligand-dependent differences in co-factor recruitment to target genes, may account for some of this variance. Our results show that suppression of COX-2 protein in response to CSE is due to the AhR, concurrent with HuR nuclear retention, both of which did not occur with classic AhR ligands, suggests divergent mechanisms of COX-2 regulation by the AhR. Despite our results showing lack of Cox-2 mRNA induction in absence of AhR expression, there is a profound increase in COX-2 protein, implicating post-transcriptional mechanisms as the way in which the AhR prevents COX-2 expression. Mammalian cells have evolved post-transcriptional mechanisms that further control inflammatory protein levels. Posttranscriptional control is accomplished by regulating nuclear export, cytoplasmic localization, translation initiation and mRNA decay, the latter being determined by the presence of the ARE in the 39-UTR of mature mRNA. Many transiently-expresses cytokines, growth factors and other mediators, including Cox-2, contain AREs and whose mRNA is rapidly destabilized. Our results demonstrate that CSE-induction of Cox2 mRNA in AhR-expressing cells was transient, and returned to baseline by 6 hours, indicative of rapid mRNA decay. Using ActD, an inhibitor of RNA synthesis, we show that when the AhR is expressed, Cox-2 mRNA is rapidly degraded. Thus, post-transcriptional regulation of protein expression by the AhR may be an important adaptive mechanism to control cellular perturbations caused by environmental stress. Stress responses can profoundly affect 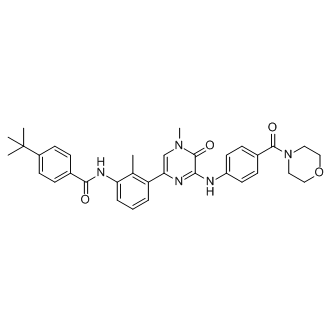 mRNA stability via the concerted efforts of numerous RNA-binding proteins including CUGBP2, TTP and HuR, all of which can play a role in regulating Cox-2 expression. Both GW786034 CUGBP2 and HuR are nuclear proteins, undergoing translocation to the cytoplasm in response to a variety of stress conditions, including c-irradiation, reactive oxygen species and ATP depletion. This nuclear-cytoplasmic shuttling is believed to provide protection against Cox-2 mRNA CX-4945 degradation. The transcriptional regulation of HuR is virtually unknown and there is no information on whether cigarette smoke alters the expression or localization of RNA-binding proteins, including CUGBP2 and HuR. Therefore, our data are the first to show that neither cigarette smoke nor AhR expression.
mRNA stability via the concerted efforts of numerous RNA-binding proteins including CUGBP2, TTP and HuR, all of which can play a role in regulating Cox-2 expression. Both GW786034 CUGBP2 and HuR are nuclear proteins, undergoing translocation to the cytoplasm in response to a variety of stress conditions, including c-irradiation, reactive oxygen species and ATP depletion. This nuclear-cytoplasmic shuttling is believed to provide protection against Cox-2 mRNA CX-4945 degradation. The transcriptional regulation of HuR is virtually unknown and there is no information on whether cigarette smoke alters the expression or localization of RNA-binding proteins, including CUGBP2 and HuR. Therefore, our data are the first to show that neither cigarette smoke nor AhR expression.