Although clinical guidelines cannot address every potential therapeutic conflict, guidance should be available for the most frequently encountered situations in which these conflicts occur, especially for situations where the potential for an adverse outcome is great when therapeutic issues are not addressed. To date, several imaging Ascomycin modalities such as echocardiography, magnetic resonance imaging and computerized tomography have been performed to measure LVH. However, ECHO might not be able to detect LVH in all dialysis patients. In addition, there are studies implied serum biomarkers like arterial vasopressin, aldosterone and cardiac Troponin T appear to predict LVH. However, stable and accurate serum biomarkers are not found, the present study evaluates whether a new biomarker for LVH can improve detection of LVH. Recent studies have demonstrated that microRNAs are present in the human circulation in a cell-free form and can be detected in circulating blood, thus may serve as a new class of blood-based biomarkers. Numerous studies reported altered plasma or serum levels of various miRNAs in patients with cardiovascular diseases, including acute myocardial infarction, myocarditis, acute and chronic heart failure and stable coronary 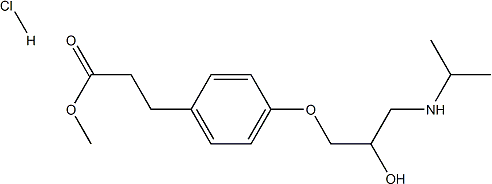 artery disease. Previous studies have found that miR-133a level was decreased in hypertrophic heart. In study presented here, we measured plasma miR-133a in MHD patients and healthy controls and Estradiol Benzoate analyzed the relationship between miR-133a level and cardiac hypertrophy. In our study, we demonstrated that circulating miR-133a level was decreased in MHD patients compared with healthy controls. Since the expression of miR-133a in heart is decreased in cardiac hypertrophy animal model, we analyzed the correlation between circulating miR-133a levels and cardiac hypertrophy in MHD patients. It was found that the miR-133a level was negatively associated with LVMI, an indicator of left ventricular hypertrophy, in these patients. The heart has capable of remodeling in response to various environmental demands and a variety of stimuli can induce it to growth or shrink. In MHD patients, primary or secondary hypertension, volume overload, hemodynamic stress and uremia toxins can alone or together induce cardiac hypertrophy. MicroRNAs are important regulators of a wide range of cellular processes by modulating gene expression and are estimated to regulate more than 30% of the genes in a cell. Recent studies both in animals and humans have demonstrated that miRNAs are present in the circulation and can be detected and quantified. Previous studies have indicated that miR-133a is highly expressed in cardiac and skeletal muscle and is regulated in hypertrophy and failure. Circulating miR-133a levels were measured in several studies and were consistently elevated in patients with cardiac infarction and were closely related with highsensitivity cardiac Troponin. In the present study, we detected circulating miR-133a levels in MHD patients and healthy controls and demonstrated that miR133a concentrations were decreased in MHD patients with LVH. Among 64 patients enrolled in our study, most of them had hypertension and about half underwent hypertension more than 5 years, but not all of them had decreased miR-133a levels. The significant association between miR-133a level and LVMI indicated that circulating miR-133a may play important role in the process of LVH and could be a biomarker of LVH.
artery disease. Previous studies have found that miR-133a level was decreased in hypertrophic heart. In study presented here, we measured plasma miR-133a in MHD patients and healthy controls and Estradiol Benzoate analyzed the relationship between miR-133a level and cardiac hypertrophy. In our study, we demonstrated that circulating miR-133a level was decreased in MHD patients compared with healthy controls. Since the expression of miR-133a in heart is decreased in cardiac hypertrophy animal model, we analyzed the correlation between circulating miR-133a levels and cardiac hypertrophy in MHD patients. It was found that the miR-133a level was negatively associated with LVMI, an indicator of left ventricular hypertrophy, in these patients. The heart has capable of remodeling in response to various environmental demands and a variety of stimuli can induce it to growth or shrink. In MHD patients, primary or secondary hypertension, volume overload, hemodynamic stress and uremia toxins can alone or together induce cardiac hypertrophy. MicroRNAs are important regulators of a wide range of cellular processes by modulating gene expression and are estimated to regulate more than 30% of the genes in a cell. Recent studies both in animals and humans have demonstrated that miRNAs are present in the circulation and can be detected and quantified. Previous studies have indicated that miR-133a is highly expressed in cardiac and skeletal muscle and is regulated in hypertrophy and failure. Circulating miR-133a levels were measured in several studies and were consistently elevated in patients with cardiac infarction and were closely related with highsensitivity cardiac Troponin. In the present study, we detected circulating miR-133a levels in MHD patients and healthy controls and demonstrated that miR133a concentrations were decreased in MHD patients with LVH. Among 64 patients enrolled in our study, most of them had hypertension and about half underwent hypertension more than 5 years, but not all of them had decreased miR-133a levels. The significant association between miR-133a level and LVMI indicated that circulating miR-133a may play important role in the process of LVH and could be a biomarker of LVH.
Month: May 2019
The overall reduction in invasion could be due to several effects synchronised invasion assay
This kinetic analysis confirmed that myoA KO Benzoylaconine parasites moved slowly for a short distance, and usually stopped after,14 mm for several minutes, before forming another semicircle, which confirmed the results of the trail deposition assay. Next, we compared invasion and replication rates between myoA KO and WT parasites. 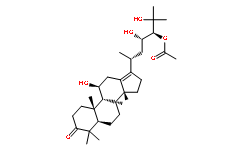 First, parasites were allowed to invade HFF cells for different times and left to replicate for 24 hours. Since one of the earliest markers of the entry process is the TJ, it was important to assess if the observed reduction in invasion rate was caused by a delay in TJ formation, or by a block in parasite progression into the host cell after TJ formation. While myoA KO parasites invaded the cell via a normally RON-shaped TJ that the majority of myoA KO parasites remained attached to the host cell without forming a TJ. Approximately 30% of myoA KO parasites formed a TJ and another 10% of all parasites were internalized. In contrast, the majority of control parasites were found to be intracellular, 10% were in the process of entry and only 10% still remained extracellular without TJ initiation. Together these data demonstrate that a step upstream of TJ formation, such as host cell recognition or reorientation of the parasites is delayed in absence of MyoA. To directly analyse the effect of MyoA depletion on host cell entry, the kinetics of this invasion step was analysed using timelapse microscopy. In total 22 entry events for the control and 27 for myoA KO parasites were compared. As expected, control parasites moved in within 20�C30 seconds in a smooth and uniform movement. In contrast, myoA KO parasites showed huge variability, with some parasites penetrating the host cell rapidly in a smooth process. However, the majority 4-(Benzyloxy)phenol entered in a spasmodic stop-and-go fashion, and appeared stalled for several seconds to minutes. The fastest recorded entry was,25 seconds, whereas the slowest entry took almost 10 minutes until the parasite was completely internalised. While these results demonstrate an important function of MyoA for efficient, smooth host cell penetration, the fact that myoA KO parasites remain capable of penetrating at a similar speed to wild-type parasites indicates that the force for host cell penetration can be generated independently of MyoA. Together these results were interpreted to be that MyoA plays an important but not essential function in multiple steps during host cell invasion. Since deletion of other components of the invasion machinery impeded mutant survival, we speculated that the function of MyoA might be partially complemented by other myosins. Together these data demonstrate that GAP45 has a role in providing the IMC with its typical structure, thereby promoting the anchorage of the MyoA motor complex in the IMC. However, since gliding motility was less affected, it appears that the parasite can efficiently produce forward movement in the absence of a myosin motor that is properly anchored along the IMC. In addition, the significant loss of invasiveness is unlikely to result from an impairment of gliding motility as previously suggested, but is rather caused by the morphological defects of these mutants. While the study of the motor mutants demonstrates that alternative pathways must be in place that can drive gliding motility and invasion, it does not rule out a critical function of other myosin motors.
First, parasites were allowed to invade HFF cells for different times and left to replicate for 24 hours. Since one of the earliest markers of the entry process is the TJ, it was important to assess if the observed reduction in invasion rate was caused by a delay in TJ formation, or by a block in parasite progression into the host cell after TJ formation. While myoA KO parasites invaded the cell via a normally RON-shaped TJ that the majority of myoA KO parasites remained attached to the host cell without forming a TJ. Approximately 30% of myoA KO parasites formed a TJ and another 10% of all parasites were internalized. In contrast, the majority of control parasites were found to be intracellular, 10% were in the process of entry and only 10% still remained extracellular without TJ initiation. Together these data demonstrate that a step upstream of TJ formation, such as host cell recognition or reorientation of the parasites is delayed in absence of MyoA. To directly analyse the effect of MyoA depletion on host cell entry, the kinetics of this invasion step was analysed using timelapse microscopy. In total 22 entry events for the control and 27 for myoA KO parasites were compared. As expected, control parasites moved in within 20�C30 seconds in a smooth and uniform movement. In contrast, myoA KO parasites showed huge variability, with some parasites penetrating the host cell rapidly in a smooth process. However, the majority 4-(Benzyloxy)phenol entered in a spasmodic stop-and-go fashion, and appeared stalled for several seconds to minutes. The fastest recorded entry was,25 seconds, whereas the slowest entry took almost 10 minutes until the parasite was completely internalised. While these results demonstrate an important function of MyoA for efficient, smooth host cell penetration, the fact that myoA KO parasites remain capable of penetrating at a similar speed to wild-type parasites indicates that the force for host cell penetration can be generated independently of MyoA. Together these results were interpreted to be that MyoA plays an important but not essential function in multiple steps during host cell invasion. Since deletion of other components of the invasion machinery impeded mutant survival, we speculated that the function of MyoA might be partially complemented by other myosins. Together these data demonstrate that GAP45 has a role in providing the IMC with its typical structure, thereby promoting the anchorage of the MyoA motor complex in the IMC. However, since gliding motility was less affected, it appears that the parasite can efficiently produce forward movement in the absence of a myosin motor that is properly anchored along the IMC. In addition, the significant loss of invasiveness is unlikely to result from an impairment of gliding motility as previously suggested, but is rather caused by the morphological defects of these mutants. While the study of the motor mutants demonstrates that alternative pathways must be in place that can drive gliding motility and invasion, it does not rule out a critical function of other myosin motors.
We assume that patients with severe PTE in acute phase have also high PTX3 levels
In other words, PTX3 may be a sensitive biomarker that indicates the ”existence of inflammation behind the disease” whereas BNP is a biomarker that reflects the severity of mechanical ventricle stress. However, the question remains on the origin of elevated PTX3 levels. A previous study on patients with left heart failure reported a Epimedoside-A negative linear correlation between PTX3 levels and ejection fraction. Leary et al. showed that higher PTX3 levels were associated with greater right ventricle mass and larger right ventricle end-diastolic volume in patients with atherosclerosis. Our study revealed a mild correlation between PTX3 levels and CO in patients with CTEPH. It is therefore possible that PTX3 is derived from inflammation in the right ventricle. On the other hand, Zabini et al. reported an increase in inflammatory cytokine concentration in PEA tissue, so it is possible that the increase in PTX3 is also produced at the organized thrombus. We could not define which tissue is the main producer of PTX3, but we speculate that there may be a complex inflammatory interaction between organized thrombi, the peripheral pulmonary vasculature, and right heart ventricle muscle. However, further investigation is needed to clarify these relationships. Regarding the Dimesna clinical diagnostic efficacies, it has previously been reported that PTX3 level in the healthy 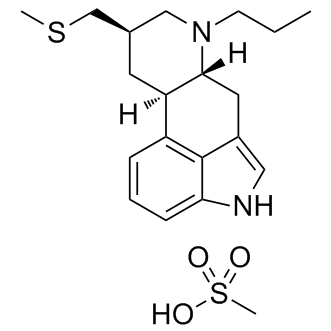 population is 2.00 ng/mL. That is similar to the PTX3 levels in the control group of this study, which consisted of patients with a history of PTE who had symptom improvement on warfarin therapy. This finding suggests that PTX3 may be a useful screening tool for identifying CTEPH even in patients with a history of PTE. For example, elevated plasma PTX3 levels in this patient population may prompt further work-up for CTEPH, which may lead to an early diagnosis. It should be noted, however, that elevated PTX3 levels have been observed in conditions other than pulmonary hypertension as described above, and careful interpretation of the data is required. Just for information, we also evaluated the PTX3 levels of three PTE patients in acute phase. They showed relatively high PTX3 levels. the elevated PTX3 level decreases as the disease gets relieved. The increased level of PTX3 in some CTEPH patients may be prolonged from the onset of acute PTE as pulmonary vasculature degeneration and/or right heart burden are prolonged. Hence, it may be difficult to distinguish the patients with severe PTE in acute phase from the patients with CTEPH. Tamura et al. previously reported elevated levels of PTX3 in patients with PAH. They found that PTX3 levels did not correlate with mPAP, PVR or BNP, which is similar to our findings. Finally, they found a relatively low level of PTX3 in patients undergoing PAH-specific treatment and suggested that PTX3 may derive from pulmonary vascular degeneration. We found that neither PAH-specific treatment nor PEA has any significant effect on PTX3 levels. It is unclear why the two studies differ in this view, but they had a common suggestion that PTX3 levels may not simply reflect the severity of PAH as described above. Our study had several limitations. First, the ROC curves may have been biased by the high prevalence of patients with CTEPH in our institute, which is considerably greater than the prevalence in most clinical settings. Second, we could not measure plasma PTX3 in a few patients who underwent PEA. There was no significant correlation between post-PEA PTX3 levels and postPEA hemodynamic parameters, and there was also no significant difference between PTX3 levels measured pre- and post-PEA. The sample size is too small to evaluate these data. Third, this study was performed retrospectively at a single institution.
population is 2.00 ng/mL. That is similar to the PTX3 levels in the control group of this study, which consisted of patients with a history of PTE who had symptom improvement on warfarin therapy. This finding suggests that PTX3 may be a useful screening tool for identifying CTEPH even in patients with a history of PTE. For example, elevated plasma PTX3 levels in this patient population may prompt further work-up for CTEPH, which may lead to an early diagnosis. It should be noted, however, that elevated PTX3 levels have been observed in conditions other than pulmonary hypertension as described above, and careful interpretation of the data is required. Just for information, we also evaluated the PTX3 levels of three PTE patients in acute phase. They showed relatively high PTX3 levels. the elevated PTX3 level decreases as the disease gets relieved. The increased level of PTX3 in some CTEPH patients may be prolonged from the onset of acute PTE as pulmonary vasculature degeneration and/or right heart burden are prolonged. Hence, it may be difficult to distinguish the patients with severe PTE in acute phase from the patients with CTEPH. Tamura et al. previously reported elevated levels of PTX3 in patients with PAH. They found that PTX3 levels did not correlate with mPAP, PVR or BNP, which is similar to our findings. Finally, they found a relatively low level of PTX3 in patients undergoing PAH-specific treatment and suggested that PTX3 may derive from pulmonary vascular degeneration. We found that neither PAH-specific treatment nor PEA has any significant effect on PTX3 levels. It is unclear why the two studies differ in this view, but they had a common suggestion that PTX3 levels may not simply reflect the severity of PAH as described above. Our study had several limitations. First, the ROC curves may have been biased by the high prevalence of patients with CTEPH in our institute, which is considerably greater than the prevalence in most clinical settings. Second, we could not measure plasma PTX3 in a few patients who underwent PEA. There was no significant correlation between post-PEA PTX3 levels and postPEA hemodynamic parameters, and there was also no significant difference between PTX3 levels measured pre- and post-PEA. The sample size is too small to evaluate these data. Third, this study was performed retrospectively at a single institution.
The significant increase in the expression of p53 mRNA in the positive steatotic or cirrhotic levels
In order to compare the efficacy of sinigrin with a common cancer drug, doxorubicin was used to treat the carcinogensinduced hepatocarcinogenesis in the rat. The results showed that after doxorubicin treatment the number of surface tumors in the rat liver was not affected whereas the group of rats that was treated with sinigrin displayed considerably reduced numbers of surface tumors. The liver weight index of doxorubicin-treated group was similar to that of the positive control group. The histological changes of liver section from rats were examined. The results indicated that there were obvious histological changes between treated and the untreated groups of rats. The liver sections from doxorubicin-treated rats displayed similar structures to those of the positive control groups of rats. These results on histological Folinic acid calcium salt pentahydrate change suggest that doxorubicin was not effective at altering carcinogen rats at this stage of tumor development. The liver sections of different control and treatment groups of rats were also analyzed with GST-p antibodies. The results ![]() of GST-p antibody labeling of the assorted liver sections provide supportive experimental evidence that rat liver functions could be restored after treatment with sinigrin relative to the negative control and the positive groups of rats. The GST-p foci showed clearly that doxorubicin treatment was ineffective managing or attenuating carcinogen-induced hepatocarcinogenesis. The levels of the rat liver enzymes ALT and AST were, importantly, significantly decreased. These data suggest that liver functions can be gradually restored after treatment with sinigrin. At the molecular level, an attempt was made to understand possible changes in gene expression associated with sinigrin treatment. The results, as indicated in Figure 9, revealed profound changes in the levels of p53 mRNA in rats following treatment with sinigrin. This change reflects the likelihood that sinigrin induces apoptosis via a p53-dependent pathway. Notably, levels of MDM2 mRNA in the sinigrin treatment group also were considerably higher than in the negative control group. This change in MDM2 expression was accompanied by alterations in the gene expression of Bax, Bcl-2 and PCNA. These findings suggest that sinigrin exerted anti-proliferative activity carcinogen-induced liver damage in the rat, and caused cell cycle arrest and amelioration of liver functions in the rat. The anti-proliferative activity of sinigrin was increased with increasing dosages. The present findings demonstrate that sinigrin is an excellent glucosinolate with medicinal properties, and add to the literature on bioactive phytochemicals in cruciferous vegetables, especially glucosinolate compounds that can inhibit the growth of liver cancer cells; such information pertaining to the anticancer activity of the glucosinolates endogenous to Brassica is limited. Sinigrin was shown to Clofentezine possess anti-cancer activities in the present study. The cell cycle analysis indicated that sinigrin caused cell cycle arrest in G0/G1 phase. Sinigrin triggered the release of cytochrome c through the down-regulation of Bcl-2 and up-regulation of Bax. The present findings reveal that Bcl-2 protein expression was significantly lower in the sinigrin treated group of rats than in the positive control group. Bax was overexpressed in the negative control and the sinigrin-treated groups relative to the positive control group. These results suggest that the over-expression of mutant p53 was increased in the rat after carcinogen exposure and subsequent carcinogenesis. The major regulator of p53 turnover, Mdm2 was found to be over-expressed at the transcriptional level in the untreated positive controls. The over-expression of Mdm2 is associated with the wild-type p53 degradation.
of GST-p antibody labeling of the assorted liver sections provide supportive experimental evidence that rat liver functions could be restored after treatment with sinigrin relative to the negative control and the positive groups of rats. The GST-p foci showed clearly that doxorubicin treatment was ineffective managing or attenuating carcinogen-induced hepatocarcinogenesis. The levels of the rat liver enzymes ALT and AST were, importantly, significantly decreased. These data suggest that liver functions can be gradually restored after treatment with sinigrin. At the molecular level, an attempt was made to understand possible changes in gene expression associated with sinigrin treatment. The results, as indicated in Figure 9, revealed profound changes in the levels of p53 mRNA in rats following treatment with sinigrin. This change reflects the likelihood that sinigrin induces apoptosis via a p53-dependent pathway. Notably, levels of MDM2 mRNA in the sinigrin treatment group also were considerably higher than in the negative control group. This change in MDM2 expression was accompanied by alterations in the gene expression of Bax, Bcl-2 and PCNA. These findings suggest that sinigrin exerted anti-proliferative activity carcinogen-induced liver damage in the rat, and caused cell cycle arrest and amelioration of liver functions in the rat. The anti-proliferative activity of sinigrin was increased with increasing dosages. The present findings demonstrate that sinigrin is an excellent glucosinolate with medicinal properties, and add to the literature on bioactive phytochemicals in cruciferous vegetables, especially glucosinolate compounds that can inhibit the growth of liver cancer cells; such information pertaining to the anticancer activity of the glucosinolates endogenous to Brassica is limited. Sinigrin was shown to Clofentezine possess anti-cancer activities in the present study. The cell cycle analysis indicated that sinigrin caused cell cycle arrest in G0/G1 phase. Sinigrin triggered the release of cytochrome c through the down-regulation of Bcl-2 and up-regulation of Bax. The present findings reveal that Bcl-2 protein expression was significantly lower in the sinigrin treated group of rats than in the positive control group. Bax was overexpressed in the negative control and the sinigrin-treated groups relative to the positive control group. These results suggest that the over-expression of mutant p53 was increased in the rat after carcinogen exposure and subsequent carcinogenesis. The major regulator of p53 turnover, Mdm2 was found to be over-expressed at the transcriptional level in the untreated positive controls. The over-expression of Mdm2 is associated with the wild-type p53 degradation.
Citrate results in lower levels of platelet-monocyte aggregates secondary to calcium
The role of platelets in lung disease is unclear, although increased platelet activation has been demonstrated in adult respiratory distress syndrome and chronic obstructive pulmonary disease. While the role of platelets in IPF is unknown, platelet trapping in the lungs of mice following intravenous bleomycin administration strongly correlated with subsequent collagen deposition, suggesting a role in fibrogenesis in this animal model. In the present study we measured markers of platelet activation in IPF patients under basal conditions and following stimulation with platelet agonists and investigated the effect of plasma from IPF patients on platelet activation. Our data demonstrate that IPF is associated with platelet hyperactivity that may be caused by the plasma environment. We have demonstrated platelet hyperactivity in patients with IPF using two mechanistically different agonists and three different markers of platelet activation, namely platelet-monocyte aggregate formation, platelet P-selectin expression, and platelet Dimesna fibrinogen binding. Increased platelet-monocyte aggregate formation is a sensitive marker of platelet activation and demonstrates a functional consequence of the increased platelet reactivity. These data provide robust evidence of increased platelet reactivity in IPF. To explore whether the hyperactivity phenotype in IPF was the result of an alteration of the platelets or their environment we used the plasma swap approach, which has been used successfully in previous studies to examine the role of plasma. Incubation of washed control platelets in IPF plasma increased platelet activation as assessed by P-selectin expression both under basal conditions and after stimulation with ADP. Platelet hyperactivity was not observed when platelets were incubated with control plasma. This indicates that the plasma environment in IPF is responsible for the observed increased platelet reactivity. Further investigation is required to identify the plasma factor or factors responsible for this effect. The two patient 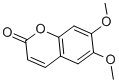 groups in this study were well matched in terms of age and gender. At the time of undertaking the platelet assays there was no evidence-based disease modifying treatment for IPF and therefore it was local practice not to treat IPF patients with immunosuppressant drugs. This is reflected in our cohort with only 1 IPF patient receiving long term low dose prednisolone. One patient in the control group was also taking long term low dose prednisolone as treatment for COPD. Although not statistically significant, there was a higher prevalence of cerebrovascular disease in the IPF group. There is a recognised association between cerebrovascular disease and platelet activation, but repeat analysis of our data following exclusion of patients with a history of cerebrovascular disease did not alter our conclusions. The platelet assays used in this study are not affected by aspirin and reanalysis of the data following exclusion of patients using antiplatelet therapies did not alter our conclusions. It has recently been demonstrated that patients with COPD have increased levels of Diperodon circulating platelet-monocyte aggregates compared to age and smoking status matched controls. The high prevalence of COPD in our control group may therefore result in the differences between the groups being underestimated, adding further support to the significance of the observed increased platelet reactivity in patients with IPF. The current smokers in this study were evenly distributed between IPF and control groups, indicating that the documented increased platelet activation and surface expression of P-selectin in smokers will not have influenced our conclusions. It is recognised that the choice of ex-vivo anticoagulant can impact platelet-monocyte association.
groups in this study were well matched in terms of age and gender. At the time of undertaking the platelet assays there was no evidence-based disease modifying treatment for IPF and therefore it was local practice not to treat IPF patients with immunosuppressant drugs. This is reflected in our cohort with only 1 IPF patient receiving long term low dose prednisolone. One patient in the control group was also taking long term low dose prednisolone as treatment for COPD. Although not statistically significant, there was a higher prevalence of cerebrovascular disease in the IPF group. There is a recognised association between cerebrovascular disease and platelet activation, but repeat analysis of our data following exclusion of patients with a history of cerebrovascular disease did not alter our conclusions. The platelet assays used in this study are not affected by aspirin and reanalysis of the data following exclusion of patients using antiplatelet therapies did not alter our conclusions. It has recently been demonstrated that patients with COPD have increased levels of Diperodon circulating platelet-monocyte aggregates compared to age and smoking status matched controls. The high prevalence of COPD in our control group may therefore result in the differences between the groups being underestimated, adding further support to the significance of the observed increased platelet reactivity in patients with IPF. The current smokers in this study were evenly distributed between IPF and control groups, indicating that the documented increased platelet activation and surface expression of P-selectin in smokers will not have influenced our conclusions. It is recognised that the choice of ex-vivo anticoagulant can impact platelet-monocyte association.