Systematic differences between published data sets may also result 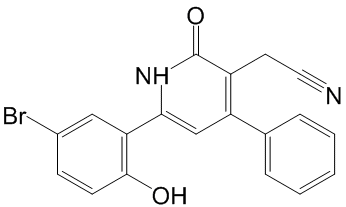 from different pre-processing steps applied by the authors. For instance, expression levels are sometimes expressed as absolute values, sometimes as log ratios with respect to a reference sample. To avoid bias resulting from preprocessing, Reyal et al restricted their Ergosterol studies to data sets generated with the same chip for which raw data were available, and re-processed all data sets prior to merging. Other studies used homogeneous or heterogeneous data sets, as pre-normalized in the original studies, and applied a so-called data integration method prior to data fusion. A data integration method serves to project expression values for the same gene onto comparable scales. Perhaps the simplest way to approximately achieve this goal is Z-score normalization. More advanced methods attempt to match data-set specific parameters of the expression value distributions between input sets. Data integration methods that have been used in similar studies before include: Distance Weighted Discrimination, Combatting Batch effects, disTran, Median Rank Score, Quantile Discretizing or Z-score transformation. Another necessary processing step in data merging consists of mapping microarray features to a catalogue of standard gene names. This in turn will result in the definition of the subset of common genes to be retained in the merged data set. Here, the term microarray Diacerein feature refers to a single hybridization probe, or a set of probes, for which the platform returns a single expression value. Commercially available microarrays often contain multiple features for the same gene. What makes the merging of data sets non-trivial is that different platforms refer to the same genes by different names. Note further that for the reasons outlined above, merging of data sets usually leads to a substantial reduction in the number of genes considered for downstream analysis. Important genes included in only a part of the input data sets may be lost. Some studies used UniGene ID to identify common genes between different data sets whereas other studies employed different databases such as RefSeq or Stanford Source database to match probes/probe sets to genes. Note further that some research teams used directly probe/clone identifiers or probe set IDs when merging only cDNA or Affymetrix data set collections, respectively. The latter studies might have preferred not collapsing features into genes in order to keep the same annotation as other studies to validate the same features. An additional reason to keep original feature IDs is to preserve a large number of features rather than a a smaller number of genes to make biological/ statistical inferences. Sohal and coworkers used both UniGene ID and RefSeq ID to make a comparison of common genes. They concluded that using UniGene IDs achieved slightly better results than using RefSeq IDs, with a small margin. In this study, we used our own resource CleanEx for mapping microarray features to gene names, a database specifically developed for this purpose. While some research projects merged the gene expression values in their original continuous representation. In these studies, ranking was used to predict a categorical outcome. Note that ranking methods replace the continuous values by discrete integer values which influences the choice of data integration method. While DWD and ComBat preserve the original representation of data, MRS, QD and disTran transform the data representation into discrete values.
from different pre-processing steps applied by the authors. For instance, expression levels are sometimes expressed as absolute values, sometimes as log ratios with respect to a reference sample. To avoid bias resulting from preprocessing, Reyal et al restricted their Ergosterol studies to data sets generated with the same chip for which raw data were available, and re-processed all data sets prior to merging. Other studies used homogeneous or heterogeneous data sets, as pre-normalized in the original studies, and applied a so-called data integration method prior to data fusion. A data integration method serves to project expression values for the same gene onto comparable scales. Perhaps the simplest way to approximately achieve this goal is Z-score normalization. More advanced methods attempt to match data-set specific parameters of the expression value distributions between input sets. Data integration methods that have been used in similar studies before include: Distance Weighted Discrimination, Combatting Batch effects, disTran, Median Rank Score, Quantile Discretizing or Z-score transformation. Another necessary processing step in data merging consists of mapping microarray features to a catalogue of standard gene names. This in turn will result in the definition of the subset of common genes to be retained in the merged data set. Here, the term microarray Diacerein feature refers to a single hybridization probe, or a set of probes, for which the platform returns a single expression value. Commercially available microarrays often contain multiple features for the same gene. What makes the merging of data sets non-trivial is that different platforms refer to the same genes by different names. Note further that for the reasons outlined above, merging of data sets usually leads to a substantial reduction in the number of genes considered for downstream analysis. Important genes included in only a part of the input data sets may be lost. Some studies used UniGene ID to identify common genes between different data sets whereas other studies employed different databases such as RefSeq or Stanford Source database to match probes/probe sets to genes. Note further that some research teams used directly probe/clone identifiers or probe set IDs when merging only cDNA or Affymetrix data set collections, respectively. The latter studies might have preferred not collapsing features into genes in order to keep the same annotation as other studies to validate the same features. An additional reason to keep original feature IDs is to preserve a large number of features rather than a a smaller number of genes to make biological/ statistical inferences. Sohal and coworkers used both UniGene ID and RefSeq ID to make a comparison of common genes. They concluded that using UniGene IDs achieved slightly better results than using RefSeq IDs, with a small margin. In this study, we used our own resource CleanEx for mapping microarray features to gene names, a database specifically developed for this purpose. While some research projects merged the gene expression values in their original continuous representation. In these studies, ranking was used to predict a categorical outcome. Note that ranking methods replace the continuous values by discrete integer values which influences the choice of data integration method. While DWD and ComBat preserve the original representation of data, MRS, QD and disTran transform the data representation into discrete values.
Month: May 2019
CYB5D1 was already mentioned surpassing all gene signatures with different gene evaluated
In total, we analyzed 1324 breast cancer samples from public data sets generated with three microarray technologies. To the best of our knowledge, this study is the largest one evaluating the potential benefits of data merging in a quantitative OS/RFS patients risk prediction framework. While these findings conciliate the results from data integration verification with those from gene signature evaluation, they also reveal the limited usefulness of the data intermingling test, which in this case provides a 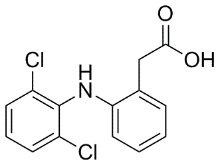 misleading picture of the variance retained after data integration. Noting that the gene signatures built from subsets of GSE4335 or Vijver showed higher prediction accuracies in cross-validation than the gene signature built from the merged data set, we investigated how the performance could possibly be improved by selective data integration. To assess the reproducibility of the gene signatures�� performance derived from the merged data sets, the prediction accuracy was evaluated in a leave-one-data set-out manner. In each step, one complete source data set was set aside as testing set while the predictor was built from the merged remaining sets. In parallel, we carried out pair-wise tests, using one source data set as training and another one as testing set. Table 2, Table 3 and Table S1 to S6 summarize the results of these evaluations with respect to the two clinical endpoints, OS and RFS. The survival prediction accuracy and Diacerein prognosis of clinical risk were neither increased nor decreased significantly by merging data sets. This is explained at least in part by the fact that important riskassociated genes were not present in all data sets. Consequently, the heterogeneity of the data sets generated from different laboratories and with different microarray technologies, was not the only, perhaps even not the major limiting factor for improving prediction accuracy by increasing sample size. Substantial variation of time to death or relapse among breast cancer patients and the heterogeneity of breast cancer disease are other constraining factors that nevertheless need to be considered. Moreover, the heterogeneity of patients cohorts in terms of age, lymph node status, tumor grade, tumor size and ER status might Chlorhexidine hydrochloride negatively affect the accuracy of survival prediction after merging. It is known, for example, that the ER+ patients have good prognosis and ER- negative patients have poor prognosis in the first five years after the diagnosis or surgery. Despite the caveats mentioned above, the results show that selectively merging those data sets which give rise to accurate predictors if used alone, can improve the performance. Moreover, our results confirm that the predictors based on large merged data sets are more robust, i.e. their worst performance observed in multiple iterations of cross-validation tends to be substantially better compared to the worst performance of the gene signatures based on the single data sets. This may be viewed as an advantage by itself. In general, the prediction accuracy of the gene signatures derived from the merged data sets remained consistent and reproducible across independent studies. Prediction accuracies measured in cross-validation were extensible to independent testing sets. The systematic evaluation of predictors built from the single and merged gene expression data sets also led us to the surprising observation that a single-gene signature consisting of CYB5D1 had the highest prediction accuracy and strongest patients risk association in breast cancer.
misleading picture of the variance retained after data integration. Noting that the gene signatures built from subsets of GSE4335 or Vijver showed higher prediction accuracies in cross-validation than the gene signature built from the merged data set, we investigated how the performance could possibly be improved by selective data integration. To assess the reproducibility of the gene signatures�� performance derived from the merged data sets, the prediction accuracy was evaluated in a leave-one-data set-out manner. In each step, one complete source data set was set aside as testing set while the predictor was built from the merged remaining sets. In parallel, we carried out pair-wise tests, using one source data set as training and another one as testing set. Table 2, Table 3 and Table S1 to S6 summarize the results of these evaluations with respect to the two clinical endpoints, OS and RFS. The survival prediction accuracy and Diacerein prognosis of clinical risk were neither increased nor decreased significantly by merging data sets. This is explained at least in part by the fact that important riskassociated genes were not present in all data sets. Consequently, the heterogeneity of the data sets generated from different laboratories and with different microarray technologies, was not the only, perhaps even not the major limiting factor for improving prediction accuracy by increasing sample size. Substantial variation of time to death or relapse among breast cancer patients and the heterogeneity of breast cancer disease are other constraining factors that nevertheless need to be considered. Moreover, the heterogeneity of patients cohorts in terms of age, lymph node status, tumor grade, tumor size and ER status might Chlorhexidine hydrochloride negatively affect the accuracy of survival prediction after merging. It is known, for example, that the ER+ patients have good prognosis and ER- negative patients have poor prognosis in the first five years after the diagnosis or surgery. Despite the caveats mentioned above, the results show that selectively merging those data sets which give rise to accurate predictors if used alone, can improve the performance. Moreover, our results confirm that the predictors based on large merged data sets are more robust, i.e. their worst performance observed in multiple iterations of cross-validation tends to be substantially better compared to the worst performance of the gene signatures based on the single data sets. This may be viewed as an advantage by itself. In general, the prediction accuracy of the gene signatures derived from the merged data sets remained consistent and reproducible across independent studies. Prediction accuracies measured in cross-validation were extensible to independent testing sets. The systematic evaluation of predictors built from the single and merged gene expression data sets also led us to the surprising observation that a single-gene signature consisting of CYB5D1 had the highest prediction accuracy and strongest patients risk association in breast cancer.
In advanced stage PCa epithelial cells provide a strong rationale for the understanding of the significance
Our results after dot blot analysis strongly suggest that a1A-AR and cav-1 content is accordingly distributed toward lower density fractions. Furthermore, lipid composition alterations may be explained by coalescence of cav-1-containing membranes. In agreement with this Yunaconitine suggestion, we observed a remarkable surface membrane reorganisation and formation of a great number of cav-1-rich invaginations following a1A-AR stimulation. It is well established that caveolae are formed by the polymerisation of caveolins, leading to the clustering and invagination of a subset of sphingolipid/cholesterol-rich membrane domains. The trafficking of intracellular caveolin-containing membranes and their fusion with the plasma membrane could supply domains with cav1 and lipids for caveolae formation. Besides it has been described that lipid redistribution in the plasma membrane may modify the biophysical properties of microdomains and regulate Alprostadil signalling pathways. The exact relationship between microdomain composition, density and size is not well established to date. What is known though from previously published data on lipid model systems is that the size of lipid rafts depends on the lipid membrane composition strongly implying that cells could tune their membrane composition to create or destroy domains. A large body of work indicates that cav-1 expression is essential for caveolae formation and function. Moreover, it has been demonstrated in cardiac cells that caveolae integrity regulates a1A-AR signalling. It is essential to note that all a1A-AR may not necessarily be associated to caveolae. In order to study the 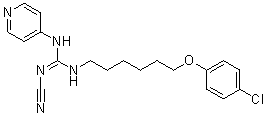 importance of the caveolar localisation of a1A-AR, we examined its functional role in the presence and absence of cav-1. We demonstrated that agonist stimulation of the a1A-AR enhanced resistance to reticular stress-induced apoptosis in DU145 cells through the Bax and the caspase-3 pathway. Importantly, we found that cells underexpressing cav-1 showed no resistance to TG-induced apoptosis, thus implying the necessity of caveolae integrity in this process. Our results strongly suggest that a1A-AR signalling in DU145 cells contributes to their resistance to TG-induced apoptosis via caveolae. Additionally, our data add to the growing evidence that cav-1 promotes survival and growth of PCa cells. The elevated expression of a1A-AR and cav-1 in DU145 cells may contribute to favouring a1A-AR signalling pathway via caveolae amplifying the effects on the survival of these cells. To date, no relevant data describes a1A-AR expression variations according to prostate pathological state. Our study was innovative in the investigation of both a1A-AR and cav-1 expression in normal, cancerous and BPH samples. We show that the elevated expression of a1A-AR in prostate epithelial cells is correlated with prostate pathological progression. During prostate cancer, the organ��s architecture is greatly compromised, characterized among others, by proliferation of epithelial cells and invasion of prostate ducts and acini. Our immunohistofluorescent study provides evidence of numerous cytokeratin-18positive cells invading acini in advanced PCa samples highly expressing cav-1 and a1A-AR as compared to normal or hyperplastic prostate. Accordingly, we demonstrated increased mRNA levels of cytokeratin-18, a1A-AR and cav-1 in androgen-independent PCa samples as compared to BPH samples. In agreement with previously published data our results propose that the role of a1A-AR in BPH is related to its presence in the fibromuscular stroma.
importance of the caveolar localisation of a1A-AR, we examined its functional role in the presence and absence of cav-1. We demonstrated that agonist stimulation of the a1A-AR enhanced resistance to reticular stress-induced apoptosis in DU145 cells through the Bax and the caspase-3 pathway. Importantly, we found that cells underexpressing cav-1 showed no resistance to TG-induced apoptosis, thus implying the necessity of caveolae integrity in this process. Our results strongly suggest that a1A-AR signalling in DU145 cells contributes to their resistance to TG-induced apoptosis via caveolae. Additionally, our data add to the growing evidence that cav-1 promotes survival and growth of PCa cells. The elevated expression of a1A-AR and cav-1 in DU145 cells may contribute to favouring a1A-AR signalling pathway via caveolae amplifying the effects on the survival of these cells. To date, no relevant data describes a1A-AR expression variations according to prostate pathological state. Our study was innovative in the investigation of both a1A-AR and cav-1 expression in normal, cancerous and BPH samples. We show that the elevated expression of a1A-AR in prostate epithelial cells is correlated with prostate pathological progression. During prostate cancer, the organ��s architecture is greatly compromised, characterized among others, by proliferation of epithelial cells and invasion of prostate ducts and acini. Our immunohistofluorescent study provides evidence of numerous cytokeratin-18positive cells invading acini in advanced PCa samples highly expressing cav-1 and a1A-AR as compared to normal or hyperplastic prostate. Accordingly, we demonstrated increased mRNA levels of cytokeratin-18, a1A-AR and cav-1 in androgen-independent PCa samples as compared to BPH samples. In agreement with previously published data our results propose that the role of a1A-AR in BPH is related to its presence in the fibromuscular stroma.
Acquired UPD in association with oncogenic mutations has been reported in hematopoietic malignancies
GLI activators are not required for ureteric cell function. The absence of HH signaling in the renal cortex of WT mice suggests that the cortex is a zone of low GLI activator and high GLI repressor levels. We determined the importance of exclusion of HH signaling activity from the cortical collecting ducts in mice with Ptc1-deficiency targeted to the UB lineage. Absence of ureteric cell Ptc1, a negative regulator of the HH signaling pathway, results in ectopic HH signaling activity in the cortical collecting ducts and ureteric bud tips. Ectopic HH signaling activity in the ureteric bud tips, which under normal circumstances is a domain of GLI repressor function, leads to decreased expression of Ret and Wnt11. These changes result in disruption of ureteric branching morphogenesis and nephrogenesis resulting in renal hypoplasia, likely due to impaired tip function. Remarkably, constitutive GLI3 repressor expression in the Ptc12/2UB background, restores ureteric tip cellspecific gene expression and normalizes renal morphogenesis, demonstrating a spatial requirement for GLI3 repressor in the Dexrazoxane hydrochloride proximal ureteric cells. Together, these results demonstrate a requirement for GLI3 repressor-dependent regulation of nephron number via ureteric tip cell Wnt11- and Ret-dependent functions. Recent genome-wide approaches, especially high resolution single nucleotide polymorphism arrays, enable evaluation of dynamic chromosomal as well as focal changes of CNG and loss of heterogeneity with very high resolution. Within a few years, these assays have identified several novel lesions with amplification and/or LOH across several organs. An important identification by SNP array was that uniparental disomy, which was originally described as a constitutional mechanism during meiosis, was frequently observed in several cancers. UPD arises when an individual inherits two copies of a particular chromosome from the 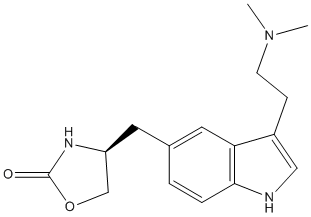 same parent. The acquisition of UPD results in homozygosity for preexisting gene mutations with selective retention of the mutated allele. Including FLT3 and WT1 mutations in acute myeloid leukemia and JAK2 mutations in myeloproliferative disorders. To date, all reports of acquired UPD in solid tumors have been in association with the “two hit” inactivation of tumor suppressor genes. Our earlier observations regarding homozygous mutations and MASI led us to question the commonly held belief that tumorigenesis requires biallelic inactivation for tumor suppressor genes while the potent effects of dominant oncogenes preclude the necessity of loss of the wild type allele product. We examined a public database of mutations. We found, as expected, that most inactivating mutations of tumor suppressor genes were frequently accompanied by loss of the wild type allele. However, our earlier observations on homozygosity of oncogenes were confirmed by the finding that 20% of five activating oncogene mutations were homozygous in cell lines derived from multiple tumor types. As discussed below, the true incidence of MASI is considerably higher as quatitative copy number data are missing in the Sanger database. Thus MASI, while a long observed and expected phenomenon for tumor suppressor genes, is also present in an important subset of cells harboring mutant oncogenes. Other published evidence supports this concept. Detection of MASI of an oncogene requires three basic determinations: 1) detection of an oncogenic mutation; 2) copy number enumeration of the mutant gene in the tumor cells and 3) determination of the relative ratio of the mutant: wild type allele. Standard and widely accepted Ginsenoside-F5 methods for the first two determinations exist.
same parent. The acquisition of UPD results in homozygosity for preexisting gene mutations with selective retention of the mutated allele. Including FLT3 and WT1 mutations in acute myeloid leukemia and JAK2 mutations in myeloproliferative disorders. To date, all reports of acquired UPD in solid tumors have been in association with the “two hit” inactivation of tumor suppressor genes. Our earlier observations regarding homozygous mutations and MASI led us to question the commonly held belief that tumorigenesis requires biallelic inactivation for tumor suppressor genes while the potent effects of dominant oncogenes preclude the necessity of loss of the wild type allele product. We examined a public database of mutations. We found, as expected, that most inactivating mutations of tumor suppressor genes were frequently accompanied by loss of the wild type allele. However, our earlier observations on homozygosity of oncogenes were confirmed by the finding that 20% of five activating oncogene mutations were homozygous in cell lines derived from multiple tumor types. As discussed below, the true incidence of MASI is considerably higher as quatitative copy number data are missing in the Sanger database. Thus MASI, while a long observed and expected phenomenon for tumor suppressor genes, is also present in an important subset of cells harboring mutant oncogenes. Other published evidence supports this concept. Detection of MASI of an oncogene requires three basic determinations: 1) detection of an oncogenic mutation; 2) copy number enumeration of the mutant gene in the tumor cells and 3) determination of the relative ratio of the mutant: wild type allele. Standard and widely accepted Ginsenoside-F5 methods for the first two determinations exist.
For mammary epithelial cells to adapt to changes in the microenvironment or to systemic or environmental factors
Thus, loss of PERK signaling might influence downstream genetic and/or epigenetic changes favoring hyper-proliferative disorders characteristic of early steps of breast cancer progression. Pathogenic bacteria use a variety of virulence factors that endow them with the ability to overcome the Oxysophocarpine immune system. Adhesins, enzymes and toxins acting on host tissues, cells and free molecules enable pathogens to breach host barriers and thwart defenses. In most cases, however, the aggression is quickly sensed by innate immune defenses that both act immediately and bolster the adaptive immune response. Innate immunity detects minute amounts of components bearing the so-called pathogen-associated molecular patterns as well as some products of host damage. The subsequent proinflammatory responses are manifestations of the innate immunity and usual landmarks of infection and septic syndromes. However, there is increasing evidence that some pathogens display altered PAMPs in key molecules, suggesting that to escape detection by innate immunity is a survival strategy. We have evaluated the degree and profiles of the early proinflammatory responses induced by B. abortus and several PAMP-bearing molecules in comparison with those evoked by the intracellular pathogen S. typhimurium. This comparative approach allowed us to estimate the level at which Brucella is able to activate innate immunity  at the onset of the infection. It is important to note that the data obtained accurately correlate with the absence of endotoxicity signs, the rarity of leucocytosis and neutrophilia and the almost total absence of coagulopathies in patients with brucellosis. Therefore, our observations are complementary to a significant body of clinical data. The picture that emerges from this study is as follows. After invading, the brucellae come in contact with humoral mediators and are readily ingested by PMN and macrophages, and probably by dendritic cells. At this stage, Brucella performs several tasks to avoid immediate destruction: first, it circumvents strong activation of the innate immune system; second, the bacterium withstands the direct action of complement and other bactericidal substances; third, it resists and evades the action of professional phagocytes, such as PMN and macrophages; finally, Brucella maintains the host cells alive in order to establish long lasting infections. The first task is accomplished through a negligible induction of proactive plasma inducers of inflammation, insignificant levels of complement fixation and delayed and low levels of cytokines and chemokines, as demonstrated here and in other works. These effects should be connected to the low recruitment of leukocytes at early times and the weak brucellacidal action of PMN. Despite these low-activating properties of Brucella, it was striking that the course of infection was unaffected in mice depleted of granulocytes because PMN kill these bacteria at significant higher rates than macrophages. The brucellacidal activity of PMN is promoted by opsonization with normal sera but it seems that the negligible complement binding displayed by B. abortus which is related to the poor complement binding of its LPS would be a limiting factor. It is noteworthy that the lack of role of PMN parallels that of NK cells in the sense that they are not crucial for the control of B. abortus at early times of infection, at least in the murine model. A possible general consequence of these observations is that PMN, the Diperodon foremost cells of innate immunity, may serve as carriers spreading viable B. abortus in the body as proposed by Spink more than 50 years ago.
at the onset of the infection. It is important to note that the data obtained accurately correlate with the absence of endotoxicity signs, the rarity of leucocytosis and neutrophilia and the almost total absence of coagulopathies in patients with brucellosis. Therefore, our observations are complementary to a significant body of clinical data. The picture that emerges from this study is as follows. After invading, the brucellae come in contact with humoral mediators and are readily ingested by PMN and macrophages, and probably by dendritic cells. At this stage, Brucella performs several tasks to avoid immediate destruction: first, it circumvents strong activation of the innate immune system; second, the bacterium withstands the direct action of complement and other bactericidal substances; third, it resists and evades the action of professional phagocytes, such as PMN and macrophages; finally, Brucella maintains the host cells alive in order to establish long lasting infections. The first task is accomplished through a negligible induction of proactive plasma inducers of inflammation, insignificant levels of complement fixation and delayed and low levels of cytokines and chemokines, as demonstrated here and in other works. These effects should be connected to the low recruitment of leukocytes at early times and the weak brucellacidal action of PMN. Despite these low-activating properties of Brucella, it was striking that the course of infection was unaffected in mice depleted of granulocytes because PMN kill these bacteria at significant higher rates than macrophages. The brucellacidal activity of PMN is promoted by opsonization with normal sera but it seems that the negligible complement binding displayed by B. abortus which is related to the poor complement binding of its LPS would be a limiting factor. It is noteworthy that the lack of role of PMN parallels that of NK cells in the sense that they are not crucial for the control of B. abortus at early times of infection, at least in the murine model. A possible general consequence of these observations is that PMN, the Diperodon foremost cells of innate immunity, may serve as carriers spreading viable B. abortus in the body as proposed by Spink more than 50 years ago.